Electronic Transitions in UV-Vis Spectroscopy: A Foundational Guide for Pharmaceutical and Biomedical Research
This article provides a comprehensive exploration of the basic theory of electronic transitions in Ultraviolet-Visible (UV-Vis) spectroscopy, tailored for researchers and drug development professionals.
Electronic Transitions in UV-Vis Spectroscopy: A Foundational Guide for Pharmaceutical and Biomedical Research
Abstract
This article provides a comprehensive exploration of the basic theory of electronic transitions in Ultraviolet-Visible (UV-Vis) spectroscopy, tailored for researchers and drug development professionals. It covers the foundational principles of how molecules absorb light, from the types of electronic transitions (σ→σ*, n→σ*, π→π*, n→π*) to the governing selection rules. The scope extends to practical methodologies for interpreting spectra, quantitative analysis using the Beer-Lambert law, and troubleshooting common experimental errors. It further addresses the critical validation of UV-Vis data through comparisons with advanced techniques like HPLC and NMR, highlighting its specific applications and limitations in a biomedical research context for tasks like compound quantification and purity assessment.
The Physics of Light Absorption: Core Principles of Electronic Transitions
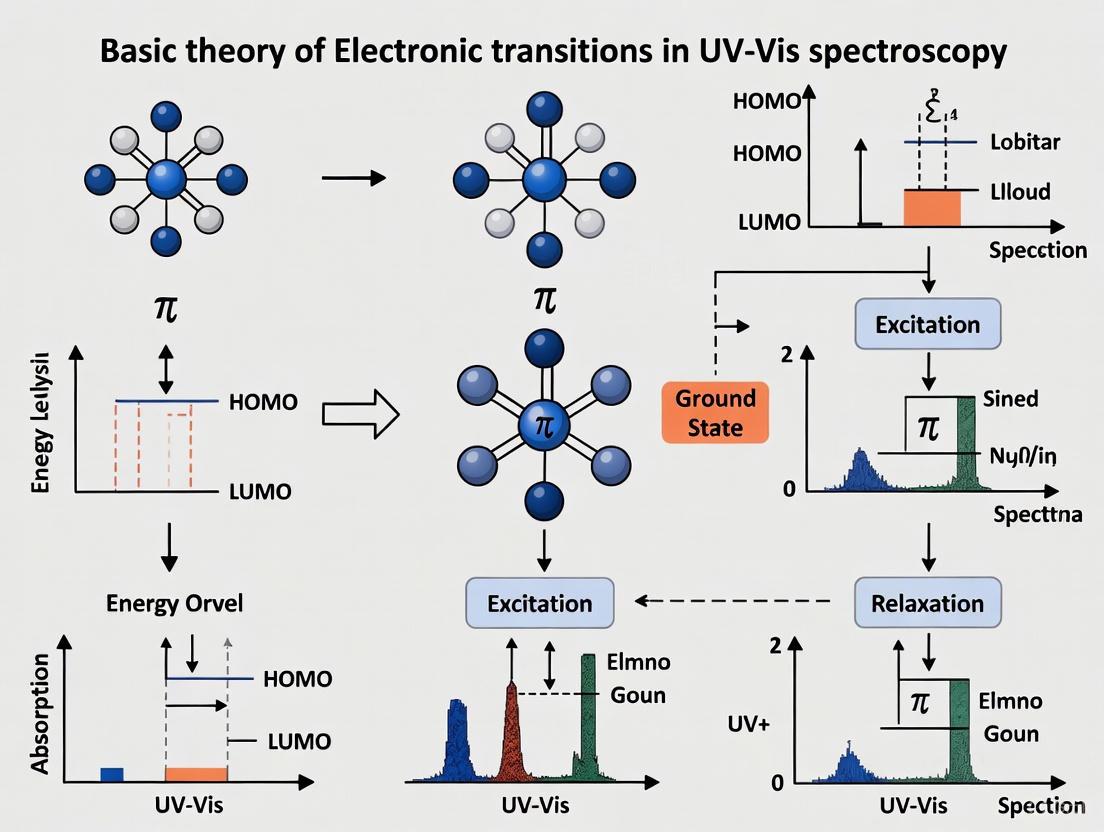
Ultraviolet-Visible (UV-Vis) spectroscopy investigates the interaction between light and matter, specifically focusing on how molecules absorb photons to undergo electronic transitions [1]. When a molecule absorbs light in the UV or visible region of the electromagnetic spectrum, electrons are promoted from their ground state to a higher energy excited state [2]. This process forms the foundation for one of the most widely used techniques in analytical chemistry, biochemistry, and pharmaceutical research for identifying compounds, determining concentrations, and understanding electronic structures [3].
The fundamental principle governing this interaction is the precise relationship between the energy of the photon absorbed and the energy gap between molecular orbitals [4]. The energy of a photon must exactly match the difference in energy between the initial and final states for absorption to occur, as described by the equation E = hν, where E is energy, h is Planck's constant, and ν is frequency [4]. This relationship connects the measurable quantity of absorbed light wavelength to the intrinsic electronic properties of molecules, providing researchers with a powerful tool for structural analysis and quantification [5].
Fundamental Physics of the Energy-Wavelength Relationship
The Quantum Mechanical Basis of Light Absorption
The interaction of light with matter occurs in discrete energy packets called photons. Each photon carries an energy quantitively described by the equation:
[
E = h\nu = \frac{hc}{\lambda}
]
where E is the energy of the photon, h is Planck's constant (6.626 × 10â»Â³â´ J·s), ν is the frequency of the light, c is the speed of light (3.00 × 10⸠m/s), and λ is the wavelength [4]. This relationship reveals the inverse proportionality between photon energy and wavelength: longer wavelengths correspond to lower energy photons, while shorter wavelengths correspond to higher energy photons [5].
When a molecule intercepts a photon whose energy precisely matches the energy difference between its ground state and an excited state (ΔE), the photon can be absorbed, promoting an electron to a higher energy orbital [1]. The frequency at which this occurs is given by:
[
\nu = \frac{ΔE}{h}
]
This fundamental quantum mechanical principle directly links the absorption wavelength observed in a UV-Vis spectrum to the electronic structure of the molecule under investigation [4].
Molecular Orbitals and Electronic Transitions
In molecular systems, electrons occupy specific molecular orbitals with defined energy levels [6]. The most critical orbitals for UV-Vis spectroscopy are:
- HOMO (Highest Occupied Molecular Orbital): The highest-energy orbital containing electrons
- LUMO (Lowest Unoccupied Molecular Orbital): The lowest-energy vacant orbital
The energy separation between the HOMO and LUMO, known as the HOMO-LUMO gap, determines the wavelength of light a molecule will absorb [6] [1]. Molecules with small HOMO-LUMO gaps absorb longer wavelengths (potentially in the visible region), while those with large gaps absorb shorter wavelengths (in the UV region) [4].
Table 1: Common Electronic Transitions in UV-Vis Spectroscopy
| Transition Type | Energy Requirement | Typical λmax Range | Molar Absorptivity (ε) | Chromophores Involved |
|---|---|---|---|---|
| σ → σ* | Very High | < 150 nm | High | C-C, C-H single bonds |
| n → σ* | High | 150-250 nm | Medium | Saturated compounds with lone pairs |
| Ï€ → Ï€* | Moderate | 200-400 nm (up to 700 nm with conjugation) | High (1,000-10,000 L·molâ»Â¹Â·cmâ»Â¹) | Alkenes, alkynes, carbonyls, conjugated systems |
| n → Ï€* | Low | 250-500 nm | Low (10-100 L·molâ»Â¹Â·cmâ»Â¹) | Compounds with both Ï€ bonds and lone pairs (e.g., carbonyls) |
For organic molecules, the most relevant transitions observed in conventional UV-Vis spectroscopy (200-800 nm) are π → π and n → π transitions [2]. These involve electrons in pi orbitals (found in double and triple bonds) and non-bonding orbitals (lone pairs on heteroatoms such as oxygen or nitrogen) [5].
Quantitative Relationship in Experimental Spectroscopy
The Beer-Lambert Law
The Beer-Lambert law forms the cornerstone of quantitative analysis using UV-Vis spectroscopy [3]. This law establishes a linear relationship between the absorbance of a solution and the concentration of the absorbing species: [ A = \varepsilon \cdot c \cdot l ] where:
Ais the measured absorbance (unitless)εis the molar absorptivity or extinction coefficient (L·molâ»Â¹Â·cmâ»Â¹)cis the concentration of the absorbing species (mol/L)lis the path length of the sample cell (cm) [1]
The molar absorptivity (ε) is a characteristic property of a compound at a specific wavelength, representing how strongly it absorbs light at that wavelength [5]. Its magnitude reflects both the size of the chromophore and the probability of the electronic transition, with strongly absorbing chromophores having values >10,000 L·molâ»Â¹Â·cmâ»Â¹ [5].
Energy Calculations in Practical Applications
The direct relationship between photon energy and wavelength allows researchers to calculate the energy associated with electronic transitions from experimental UV-Vis spectra. The energy difference between molecular orbitals can be determined using the absorption maximum (λmax):
[
ΔE = \frac{hc}{\lambda_{max}}
]
where λmax is the wavelength of maximum absorption [4].
Table 2: Energy-Wavelength Conversion for Common Electronic Transitions
| Compound | Transition Type | λmax (nm) | Energy (kJ/mol) | Energy (kcal/mol) | Orbitals Involved |
|---|---|---|---|---|---|
| Ethene | π → π* | 170 | 704 | 164 | HOMO(π) → LUMO(π*) |
| Butadiene | π → π* | 217 | 551 | 132 | HOMO(π) → LUMO(π*) |
| Hexatriene | π → π* | 258 | 464 | 111 | HOMO(π) → LUMO(π*) |
| Carbonyl n→π* | n → π* | 290 | 412 | 98 | HOMO(n) → LUMO(π*) |
| Carbonyl π→π* | π → π* | 180 | 664 | 159 | HOMO(π) → LUMO(π*) |
| β-Carotene | π → π* | 470 | 254 | 61 | HOMO(π) → LUMO(π*) |
This quantitative relationship enables researchers to predict the color of compounds, understand bond strengths, and design molecules with specific light-absorption properties for applications ranging from photovoltaics to pharmaceuticals [4].
Experimental Protocols for Validating the Energy-Wavelength Relationship
Protocol 1: Determining λmax and Molar Absorptivity
Objective: To determine the wavelength of maximum absorption (λmax) and molar absorptivity (ε) for a conjugated organic compound.
Materials and Reagents:
- UV-Vis spectrophotometer with scanning capability
- Matched quartz cuvettes (path length typically 1.0 cm)
- Analytical balance
- Volumetric flasks
- HPLC-grade solvent (e.g., hexane, methanol, water)
- Pure analyte of known molecular weight
Procedure:
- Sample Preparation: Precisely weigh 5-10 mg of analyte and dissolve in solvent to make a stock solution of known concentration (typically 10â»Â³ to 10â»âµ M).
- Dilution Series: Prepare a series of dilutions covering an appropriate concentration range (e.g., 10â»â´ to 10â»â¶ M).
- Blank Measurement: Fill a cuvette with pure solvent and place in the reference beam.
- Spectral Scan: Place the most concentrated sample in the sample beam and scan from 200-800 nm (or appropriate range) to identify λmax.
- Absorbance Measurement: At the identified λmax, measure the absorbance of all standard solutions.
- Data Analysis: Plot absorbance versus concentration. The slope of this plot equals
ε·l, from which ε can be calculated using the known path length.
Validation: The linearity of the Beer-Lambert plot (R² > 0.995) confirms the validity of the relationship within the concentration range studied [3].
Protocol 2: Investigating Conjugation Effects on λmax
Objective: To demonstrate how extended conjugation reduces the HOMO-LUMO gap, shifting λmax to longer wavelengths.
Materials and Reagents:
- Series of polyenes with increasing conjugation length (e.g., ethene, butadiene, hexatriene, octatetraene)
- UV-grade hexane as solvent
- UV-Vis spectrophotometer
- Quartz cuvettes
Procedure:
- Solution Preparation: Prepare solutions of each polyene at approximately 10â»âµ M concentration in hexane.
- Spectral Acquisition: Obtain full UV-Vis spectra for each compound.
- λmax Determination: Identify the wavelength of maximum absorption for each polyene.
- Energy Calculation: Calculate the energy difference (ΔE) for each transition using the equation
ΔE = hc/λmax. - Data Correlation: Plot λmax versus number of double bonds and ΔE versus number of double bonds.
Expected Results: As conjugation increases, λmax shifts to longer wavelengths (bathochromic shift), and ΔE decreases, demonstrating the relationship between molecular structure, orbital energies, and absorption wavelength [4].
Factors Influencing the Energy-Wavelength Relationship
Chromophore Structure and Conjugation
Conjugation represents the most significant structural factor affecting the energy-wavelength relationship in organic molecules [5]. Extended conjugation across multiple double bonds leads to a bathochromic shift (red shift), where absorption moves to longer wavelengths [4]. This occurs because conjugation lowers the energy of the π* orbital while raising the energy of the π orbital, thereby reducing the HOMO-LUMO gap [6].
For example, while isolated ethene absorbs at 170 nm, butadiene with two conjugated double bonds absorbs at 217 nm, and hexatriene with three conjugated double bonds absorbs at 258 nm [4]. With sufficient conjugation, absorption moves into the visible region, producing colored compounds such as β-carotene (λmax = 470 nm), which appears orange [1].
Solvent Effects
The solvent environment significantly influences absorption spectra through various interactions [2]. The same compound may exhibit different λmax values in different solvents due to:
- Polarity Effects: Polar solvents tend to cause bathochromic (red) shifts for π→π* transitions due to better stabilization of the more polar excited state [2].
- Hydrogen Bonding: For n→π* transitions, hydrogen-bonding solvents (e.g., water, alcohols) stabilize the non-bonding electrons, increasing the transition energy and causing a hypsochromic shift (blue shift) to shorter wavelengths [2].
- Polarizability: Solvents with high electron polarizability can differentially stabilize ground and excited states through dispersion forces.
These solvent effects must be carefully controlled and reported for reproducible results, with solvent choice specified in all experimental protocols [3].
Auxochromic Substituents
Auxochromes are functional groups that, while not chromophores themselves, modify the absorption characteristics of chromophores when attached to them [6]. Common auxochromes include -OH, -NH₂, -OR, and -Cl groups, which typically contain lone pairs of electrons that can interact with the π system of the chromophore [6].
The effect of auxochromes depends on their electronic character:
- Electron-donating groups (e.g., -OH, -NHâ‚‚) typically cause bathochromic shifts by raising the HOMO energy level
- Electron-withdrawing groups (e.g., -NOâ‚‚, -COOH) may cause hypsochromic or bathochromic shifts depending on their position and interaction with the chromophore
These substituent effects follow predictable patterns that have been codified in empirical rules such as the Woodward-Fieser rules for dienes and carbonyl compounds [3].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential Research Reagents and Materials for UV-Vis Spectroscopy
| Item | Function/Application | Technical Specifications | Importance in Energy-Wavelength Studies |
|---|---|---|---|
| Quartz Cuvettes | Sample holder for UV-Vis measurements | Path lengths: 1.0 cm (standard); Transmission range: 200-2500 nm | Essential for accurate absorbance measurements; Glass absorbs UV light, so quartz is necessary below 350 nm |
| UV-Vis Spectrophotometer | Instrument for measuring light absorption | Wavelength range: 190-1100 nm; Bandwidth: <2 nm for research-grade | Must provide precise wavelength control to accurately determine λmax and energy calculations |
| HPLC-Grade Solvents | Dissolving samples for analysis | Low UV absorbance; High purity | Minimize solvent background absorption; Common choices: water, acetonitrile, hexane, methanol |
| Deuterium Lamp | UV light source for spectrophotometer | Covers 190-400 nm range | Provides continuous spectrum in UV region for electronic transition studies |
| Tungsten-Halogen Lamp | Visible light source for spectrophotometer | Covers 350-1100 nm range | Essential for studying transitions in visible region (colored compounds) |
| NIST-Traceable Standards | Wavelength and absorbance calibration | Holmium oxide filters (wavelength); Neutral density filters (absorbance) | Ensure instrument accuracy for valid energy-wavelength relationship studies |
| Microvolume UV-Vis Systems | Sample-limited applications | Requires 0.5-2 µL sample volume; Path length: 0.2-1.0 mm | Enables analysis of precious samples while maintaining accurate energy-wavelength measurements |
| Nispomeben | Nispomeben, CAS:1443133-41-2, MF:C21H27NO4, MW:357.4 g/mol | Chemical Reagent | Bench Chemicals |
| Janex-1-m | Janex-1-m, CAS:406484-24-0, MF:C15H13N3O3, MW:283.28 g/mol | Chemical Reagent | Bench Chemicals |
Advanced Considerations and Research Applications
Instrumental Factors Affecting Measurements
Several instrumental parameters must be controlled to obtain accurate data for energy-wavelength relationship studies [3]:
- Spectral Bandwidth: The range of wavelengths transmitted simultaneously affects resolution. For sharp absorption peaks, narrower bandwidths (1-2 nm) are essential to accurately determine λmax [3].
- Stray Light: Light reaching the detector at wavelengths other than the target wavelength causes deviation from the Beer-Lambert law, particularly at high absorbances (>2 AU) [3].
- Wavelength Accuracy: Small errors in wavelength calibration become significant when calculating transition energies, especially for sharp peaks [3].
Pharmaceutical and Biopharmaceutical Applications
In pharmaceutical research, understanding the energy-wavelength relationship enables critical quality control and characterization applications [7]:
- Drug Purity Assessment: UV-Vis spectroscopy quantifies API (Active Pharmaceutical Ingredient) concentration and detects impurities based on their characteristic absorption spectra [7].
- Protein Characterization: Proteins containing aromatic amino acids (tryptophan, tyrosine, phenylalanine) exhibit UV absorption at 280 nm, allowing quantification without separation [3].
- Real-Time Release Testing (RTRT): UV-Vis spectroscopy serves as a process analytical technology (PAT) tool for real-time quality assessment during manufacturing, with studies confirming sufficient penetration depth (up to 1.38 mm) for representative sampling in tablet analysis [7].
- Binding Studies: Changes in absorption spectra upon ligand binding provide information about protein-ligand interactions and binding constants.
The precise relationship between absorption characteristics and molecular structure makes UV-Vis spectroscopy an indispensable tool throughout drug development, from initial discovery to final quality control [7].
The fundamental relationship between photon energy and wavelength provides the physical basis for interpreting electronic transitions in UV-Vis spectroscopy. Through the direct correlation E = hc/λ, researchers can connect measurable absorption wavelengths to molecular orbital energy gaps, enabling both qualitative identification and quantitative analysis of chemical compounds. Factors including chromophore conjugation, solvent effects, and auxochromic substituents systematically influence this relationship, providing valuable structural information. In pharmaceutical applications, these principles support critical quality assessments and real-time monitoring, demonstrating the enduring utility of the energy-wavelength relationship in advancing scientific research and drug development.
Ultraviolet-Visible (UV-Vis) spectroscopy is a fundamental analytical technique that probes the electronic structure of molecules by measuring their absorption of light in the ultraviolet and visible regions of the electromagnetic spectrum (typically 190-800 nm) [8] [9]. The core principle underlying this technique is that molecules contain electrons in specific molecular orbitals, and these electrons can be promoted to higher energy orbitals when they absorb photons with energy matching the difference between orbital energy levels [4]. This absorption of light results in electronic transitions, which provide crucial information about molecular structure, conjugation, and functional groups [10] [11].
The energy required for electronic transitions follows Planck's relation (E = hν), where the energy gap (ΔE) between molecular orbitals determines the wavelength of light absorbed [4] [12]. In molecular orbital theory, electrons normally occupy the lowest energy orbitals in the ground state configuration. When light of appropriate energy interacts with a molecule, electrons may be excited from the highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO), or to other higher energy unoccupied orbitals [13] [5]. This promotion of electrons from ground state to excited state orbitals forms the basis for understanding the four primary types of electronic transitions in organic molecules: σ→σ, n→σ, π→π, and n→π transitions [12] [11].
Fundamental Theory of Molecular Orbitals and Transitions
Molecular Orbital Framework
In molecular orbital theory, atomic orbitals combine to form molecular orbitals that are delocalized over the entire molecule [14]. When atoms bond together, their atomic orbitals interact to create bonding orbitals (lower energy) and antibonding orbitals (higher energy). The bonding orbitals are characterized by constructive interference between atomic wavefunctions, with electron density concentrated between nuclei, while antibonding orbitals result from destructive interference, with electron density excluded from the internuclear region [14] [4]. The sigma (σ) orbitals are formed by head-on overlap of atomic orbitals, creating electron density along the bond axis. Pi (π) orbitals result from side-by-side overlap of p orbitals, creating electron density above and below the bond axis. Non-bonding (n) orbitals are typically lone pair orbitals on heteroatoms that do not participate in bonding [15].
The energy ordering of these orbitals generally follows: σ < π < n < π* < σ* [15]. This energy hierarchy is crucial for understanding the relative energies required for different electronic transitions. When molecules absorb ultraviolet or visible light, electrons are promoted from filled to empty orbitals, with the energy requirement determined by the difference between these orbital energy levels [13].
Selection Rules and Transition Probabilities
Not all possible electronic transitions occur with equal probability. Selection rules govern the likelihood of transitions between electronic states [11]. The spin selection rule states that transitions between states of different spin multiplicity are forbidden—singlet-to-singlet transitions are allowed, while singlet-to-triplet transitions are forbidden. The Laporte selection rule (or parity selection rule) states that for centrosymmetric molecules, transitions between orbitals of the same parity (gg or uu) are forbidden, while transitions between orbitals of different parity (gu) are allowed [11].
The intensity of absorption is proportional to the square of the transition dipole moment, which depends on the overlap between the orbitals involved in the transition [5]. Forbidden transitions may still occur with weak intensity due to vibronic coupling, which relaxes the strict selection rules by mixing vibrational and electronic states [11].
The Four Primary Electronic Transitions
σ→σ* Transitions
Sigma to sigma-star transitions represent the promotion of an electron from a bonding σ orbital to an antibonding σ* orbital [13] [12]. These transitions require substantial energy since σ bonds are strong, typically resulting in absorption at short wavelengths in the vacuum UV region below 150 nm [4]. For example, molecular hydrogen (H₂) undergoes a σ→σ* transition at approximately 111-135 nm, corresponding to an energy gap of about 258 kcal/mol [13] [14]. Similarly, ethane exhibits a σ→σ* transition around 135 nm [12]. These high-energy transitions are generally not observable with standard UV-Vis spectrophotometers, which typically operate down to about 190-220 nm [13] [4]. The study of σ→σ* transitions requires specialized instrumentation, including vacuum chambers to eliminate oxygen absorption and high-energy light sources [4].
Table 1: Characteristics of σ→σ* Transitions
| Compound | λmax (nm) | Energy (kcal/mol) | ε (L·molâ»Â¹Â·cmâ»Â¹) | Observation Requirements |
|---|---|---|---|---|
| Hâ‚‚ (Hydrogen) | 111-135 | ~258 | - | Vacuum UV conditions |
| CH₃-CH₃ (Ethane) | 135 | ~212 | - | Vacuum UV conditions |
| C-H bond | <120 | >238 | - | Vacuum UV conditions |
| C-C bond | <120 | >238 | - | Vacuum UV conditions |
n→σ* Transitions
Non-bonding to sigma-star transitions occur when electrons in non-bonding orbitals (lone pairs) on heteroatoms such as oxygen, nitrogen, sulfur, or halogens are promoted to σ* orbitals [12] [11]. These transitions generally require less energy than σ→σ* transitions, typically appearing in the range of 150-250 nm [11]. For instance, water exhibits an n→σ* transition at 167 nm with a molar absorptivity of 7,000 L·molâ»Â¹Â·cmâ»Â¹ [12]. Similarly, methanol and other alcohols show n→σ* transitions below 200 nm [5]. The exact energy of n→σ* transitions depends on the heteroatom involved and the electron-withdrawing or donating characteristics of substituents. These transitions are typically weaker than π→π* transitions but stronger than n→π* transitions [11].
Table 2: Characteristics of n→σ* Transitions
| Compound | λmax (nm) | ε (L·molâ»Â¹Â·cmâ»Â¹) | Heteroatom | Solvent |
|---|---|---|---|---|
| Hâ‚‚O (Water) | 167 | 7,000 | Oxygen | - |
| CH₃OH (Methanol) | 183 | - | Oxygen | - |
| CH₃Cl | 173 | - | Chlorine | - |
| (CH₃)₂O (Dimethyl ether) | 184 | - | Oxygen | - |
π→π* Transitions
Pi to pi-star transitions involve the promotion of an electron from a bonding Ï€ orbital to an antibonding Ï€* orbital in systems with double bonds or aromatic rings [13] [12]. Isolated Ï€ bonds, such as in ethylene (ethene), absorb at around 165-174 nm [13] [4]. However, conjugation dramatically affects the energy of π→π* transitions—as conjugation increases, the energy gap between Ï€ and Ï€* orbitals decreases, resulting in absorption at longer wavelengths [13] [10]. For example, 1,3-butadiene absorbs at 217 nm, and 1,3,5-hexatriene at 258 nm [13]. With extensive conjugation, π→π* transitions can shift into the visible region, as evidenced by β-carotene (11 conjugated double bonds) which absorbs at 470 nm, appearing orange [13] [4]. These transitions are typically strong, with high molar absorptivities (ε > 10,000 L·molâ»Â¹Â·cmâ»Â¹) due to good orbital overlap between the Ï€ and Ï€* orbitals [5] [15].
Table 3: Characteristics of π→π* Transitions in Selected Alkenes
| Compound | λmax (nm) | ε (L·molâ»Â¹Â·cmâ»Â¹) | Conjugation Length | Color |
|---|---|---|---|---|
| Ethene | 165-174 | - | Isolated π bond | Colorless |
| 1,3-Butadiene | 217 | 21,000 | 2 conjugated π bonds | Colorless |
| 1,3,5-Hexatriene | 258 | 35,000 | 3 conjugated π bonds | Colorless |
| β-Carotene | 470 | 15,000 | 11 conjugated π bonds | Orange |
n→π* Transitions
Non-bonding to pi-star transitions occur in molecules containing both Ï€ bonds and heteroatoms with non-bonding electrons, such as carbonyl compounds (aldehydes, ketones, carboxylic acids) and azo compounds [13] [15]. In these transitions, an electron from a non-bonding orbital on a heteroatom (typically oxygen or nitrogen) is promoted to a Ï€* orbital [15]. These are the lowest energy electronic transitions, typically occurring in the range of 270-300 nm for simple carbonyls [15] [10]. For example, acetone exhibits an n→π* transition at 275 nm [15]. These transitions are characteristically weak (ε = 10-100 L·molâ»Â¹Â·cmâ»Â¹) due to poor orbital overlap between the non-bonding orbital (which is perpendicular to the Ï€ system) and the Ï€* orbital [5] [15]. The n→π* transitions are highly sensitive to solvent effects, typically shifting to shorter wavelengths (hypsochromic or blue shift) with increasing solvent polarity [12] [15].
Table 4: Characteristics of n→π* Transitions
| Compound | λmax (nm) | ε (L·molâ»Â¹Â·cmâ»Â¹) | Functional Group | Transition Type |
|---|---|---|---|---|
| Acetone | 275 | ~15 | Ketone | n→π* |
| Acetaldehyde | 290 | ~17 | Aldehyde | n→π* |
| 4-methyl-3-penten-2-one | 314 | - | Enone | n→π* (conjugated) |
Experimental Protocols in UV-Vis Spectroscopy
Instrumentation and Measurement Methodology
UV-Vis spectroscopy relies on sophisticated instrumentation to accurately measure light absorption across the ultraviolet and visible spectrum [8]. A typical UV-Vis spectrophotometer consists of four main components: a light source, wavelength selector, sample container, and detector [8] [9]. For measurements across both UV and visible regions, instruments often employ multiple light sources—deuterium lamps for UV light (190-400 nm) and tungsten or halogen lamps for visible light (400-800 nm) [8]. The wavelength selector, typically a monochromator containing a diffraction grating, isolates specific wavelengths from the broad spectrum emitted by the light source [8]. Modern monochromators generally feature diffraction gratings with 1200-2000 grooves per mm, providing optimal resolution for spectroscopic measurements [8].
Sample containers (cuvettes) must be carefully selected based on the wavelength range of interest. Quartz or fused silica cuvettes are essential for UV measurements below 350 nm, as glass and plastic cuvettes absorb strongly in the UV region [8]. For measurements in the visible range only, glass or plastic cuvettes may be suitable. The path length of cuvettes is typically 1 cm, though shorter path lengths (e.g., 1 mm) may be used for highly absorbing samples [8]. The detector, often a photomultiplier tube (PMT) or photodiode, converts the transmitted light intensity into an electrical signal for measurement [8].
The fundamental measurement in UV-Vis spectroscopy follows the Beer-Lambert Law: A = εlc, where A is absorbance, ε is the molar absorptivity coefficient (L·molâ»Â¹Â·cmâ»Â¹), l is the path length (cm), and c is the concentration (mol·Lâ»Â¹) [8] [10]. Absorbance values between 0.1 and 1.0 are generally considered optimal for accurate quantification, corresponding to 10-80% light absorption [8].
Sample Preparation and Solvent Selection
Proper sample preparation is critical for obtaining reliable UV-Vis spectra. Samples are typically prepared as solutions, with concentrations carefully chosen to ensure absorbance values remain within the instrument's linear dynamic range (generally A < 1) [8]. For quantitative work, a series of standard solutions with known concentrations is prepared to establish a calibration curve [8].
Solvent selection requires careful consideration as solvents can significantly influence absorption spectra. Common solvents for UV-Vis spectroscopy include [5]:
- Water: Transparent above 190 nm, suitable for aqueous samples
- Hexane/cyclohexane: Non-polar, transparent above 200 nm
- Acetonitrile: Polar aprotic, transparent above 190 nm
- Methanol/ethanol: Polar protic, transparent above 205 nm
- Chloroform: Transparent above 245 nm
Solvent effects are particularly important for n→π* transitions, which typically exhibit blue shifts (hypsochromic shifts) in polar solvents due to hydrogen bonding with lone pair electrons [12] [15]. Conversely, π→π* transitions often show red shifts (bathochromic shifts) in polar solvents due to stabilization of the excited state [11].
Table 5: UV Cutoff Wavelengths of Common Solvents
| Solvent | UV Cutoff (nm) | Polarity | Suitability for Different Transitions |
|---|---|---|---|
| Water | 190 | High | Good for most transitions |
| Acetonitrile | 190 | Medium-High | Excellent for most transitions |
| Hexane | 200 | Low | Good for most transitions |
| Methanol | 205 | High | Good for most transitions |
| Ethanol | 205 | High | Good for most transitions |
| Chloroform | 245 | Low | Limited for n→π* transitions |
| Carbon tetrachloride | 265 | Low | Limited for n→π* transitions |
Data Collection and Analysis Protocol
Standard protocol for collecting and interpreting UV-Vis spectral data [8] [10]:
- Instrument Calibration: Warm up the spectrophotometer for 15-30 minutes to stabilize the light source. Perform a baseline correction with the blank solvent to account for solvent absorption.
- Spectral Scanning: Scan samples over the appropriate wavelength range (typically 190-800 nm) with a scanning speed appropriate for the sample (typically medium speed for routine analyses).
- Peak Identification: Identify λmax values (wavelengths of maximum absorption) for all significant peaks in the spectrum.
- Absorbance Measurement: Record absorbance values at each λmax, ensuring they fall within the optimal range of 0.1-1.0. For concentrated samples, apply appropriate dilution factors.
- Spectral Interpretation: Correlate observed transitions with molecular structure:
- Absorbance at 200-220 nm suggests isolated π bonds or n→σ* transitions
- Absorbance at 210-250 nm indicates conjugated dienes or π→π* transitions
- Absorbance at 250-300 nm suggests extended conjugation or aromatic systems
- Weak absorbance at 270-300 nm indicates n→π* transitions in carbonyls
- Quantitative Analysis: For concentration determination, measure absorbance at the specific λmax and apply the Beer-Lambert law using previously determined molar absorptivity values.
Advanced Considerations and Research Applications
Chromophores and Conjugation Effects
Chromophores are functional groups or structural elements that absorb specific wavelengths of UV or visible light due to the presence of π electrons or heteroatoms with non-bonding electrons [13] [5]. Common chromophores include carbonyl groups, carbon-carbon double and triple bonds, aromatic rings, azo groups (-N=N-), and nitro groups (-NO₂) [5] [15]. The absorption characteristics of chromophores are dramatically influenced by conjugation—the presence of alternating single and multiple bonds that creates a system of delocalized π electrons [13] [10].
Conjugation reduces the energy gap between π and π* orbitals, resulting in bathochromic shifts (red shifts) to longer wavelengths and hyperchromic effects (increased absorption intensity) [13] [4]. This effect is clearly demonstrated in polyene series: ethene (λmax = 170 nm), 1,3-butadiene (λmax = 217 nm), 1,3,5-hexatriene (λmax = 258 nm), and β-carotene with 11 conjugated double bonds (λmax = 470 nm) [13] [4]. In drug development, understanding conjugation effects is crucial for designing molecules with specific light absorption properties, which can influence photostability, color, and biological activity [8] [9].
Solvent Effects and Spectral Shifts
Solvent choice significantly impacts UV-Vis spectra through several mechanisms [12] [11]. Polar solvents can cause bathochromic (red) shifts in π→π* transitions by stabilizing the more polar excited state through dipole-dipole interactions [11]. Conversely, n→π* transitions typically exhibit hypsochromic (blue) shifts in polar protic solvents due to hydrogen bonding with the non-bonding electrons in the ground state, which increases the energy required for transition [15] [11]. These solvent effects must be carefully considered when comparing literature values, as λmax can shift by 10-30 nm depending on solvent polarity [12].
Quantitative Analysis and the Beer-Lambert Law
The Beer-Lambert law forms the foundation for quantitative applications of UV-Vis spectroscopy [8] [10]. According to this relationship, absorbance (A) is directly proportional to concentration (c), path length (l), and the molar absorptivity coefficient (ε): A = εlc [8]. For accurate quantification, measurements should be made at the wavelength of maximum absorption (λmax), and absorbance values should fall within the linear range of the instrument (typically A < 1) [8]. The molar absorptivity (ε) provides information about transition probability, with values ranging from <100 for forbidden transitions (n→π) to >10,000 for allowed transitions (π→π in conjugated systems) [5] [15].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 6: Essential Research Reagents and Materials for UV-Vis Spectroscopy
| Item | Specification/Type | Function/Application | Key Considerations |
|---|---|---|---|
| Solvents | HPLC-grade hexane, acetonitrile, methanol, water | Sample dissolution and reference measurements | UV transparency, purity, compatibility with sample |
| Cuvettes | Quartz (UV), glass (vis), plastic (vis) | Sample containment during measurement | Path length (typically 1 cm), wavelength compatibility |
| Light Sources | Deuterium lamp (UV), tungsten/halogen lamp (vis) | Provide broad-spectrum illumination | Stability, lifespan, switching at 300-350 nm |
| Wavelength Selector | Monochromator with diffraction grating | Isolate specific wavelengths | Groove density (1200-2000 grooves/mm), resolution |
| Detectors | Photomultiplier tube (PMT), photodiode | Convert light intensity to electrical signal | Sensitivity, dynamic range, signal-to-noise ratio |
| Standard Compounds | Potassium dichromate, holmium oxide | Instrument calibration and validation | Wavelength accuracy verification, absorbance standards |
| Buffer Systems | Phosphate, acetate, borate buffers | Maintain pH for biological samples | UV transparency at working concentrations |
| Reference Materials | Solvent-matched blanks | Baseline correction | Matrix matching with sample solution |
| UMPK ligand 1 | UMPK ligand 1, MF:C15H22N4O5S, MW:370.4 g/mol | Chemical Reagent | Bench Chemicals |
| 8-Nitro-2'3'cAMP | 8-Nitro-2'3'cAMP, MF:C10H11N6O8P, MW:374.20 g/mol | Chemical Reagent | Bench Chemicals |
The comprehensive understanding of σ→σ, n→σ, π→π, and n→π transitions provides a fundamental framework for interpreting UV-Vis spectra and extracting meaningful structural information about molecules. These electronic transitions, governed by well-defined selection rules and influenced by molecular structure, conjugation, and solvent environment, serve as powerful diagnostic tools in molecular characterization [13] [12] [10]. The experimental protocols outlined in this work, combined with proper instrumentation and reagent selection, enable researchers to obtain reliable, reproducible spectral data for both qualitative identification and quantitative analysis [8] [9].
In pharmaceutical research and drug development, UV-Vis spectroscopy offers invaluable applications ranging from compound identification and purity assessment to concentration determination and kinetic studies [8] [9]. The ability to correlate spectral features with specific electronic transitions allows medicinal chemists to make informed decisions about molecular design, particularly regarding chromophore incorporation and conjugation extension to modulate light absorption properties. As UV-Vis instrumentation continues to advance with improved sensitivity, miniaturization, and automation, the technique remains an indispensable tool in the scientist's analytical arsenal, providing immediate insights into electronic structure through the well-established principles of orbital interactions.
Understanding Charge-Transfer Transitions in Metal Complexes and Drug Molecules
Charge-transfer (CT) transitions represent a crucial class of electronic transitions in molecular spectroscopy, playing a fundamental role in ultraviolet-visible (UV-Vis) research. These transitions occur when electronic charge is redistributed between different parts of a molecular system, typically between an electron donor and an electron acceptor. In contrast to localized d-d transitions in metal complexes, CT transitions involve significant spatial redistribution of electron density and are characterized by high intensity and broad spectral bands [16] [17]. The fundamental theory underlying these transitions was first comprehensively described by Mulliken in the 1950s, establishing the quantum mechanical basis for donor-acceptor interactions [18].
In the context of metal complexes, CT transitions can be classified into two primary categories: Ligand-to-Metal Charge Transfer (LMCT), where electron density moves from ligand-based orbitals to metal-centered orbitals, and Metal-to-Ligand Charge Transfer (MLCT), involving electron transfer from metal-based orbitals to ligand-based orbitals [19] [16]. The directionality of these transitions depends critically on the relative energies of the molecular orbitals involved and the redox properties of both the metal center and ligands [16]. Understanding these electronic transitions provides fundamental insights into photophysical processes, redox chemistry, and biological recognition events relevant to pharmaceutical development.
Fundamental Mechanisms and Theoretical Framework
Electronic Structure Basis
Charge-transfer transitions originate from the quantum mechanical interaction between electron donors and acceptors, forming new molecular aggregates in the ground state known as charge-transfer complexes (CTCs) [18]. These complexes exhibit unique electronic structures characterized by:
- Orbital Interactions: CT transitions typically involve the highest occupied molecular orbital (HOMO) of the donor and the lowest unoccupied molecular orbital (LUMO) of the acceptor [18]. The energy gap between these orbitals determines the wavelength of the CT absorption band.
- Transition Intensity: CT bands are notably intense (high molar absorptivity) because they involve orbitals that are spatially separated, resulting in large transition dipole moments [17]. This contrasts with forbidden d-d transitions that yield weak absorption bands.
- Solvent Effects: The position and intensity of CT bands are highly sensitive to solvent polarity due to stabilization of the charge-separated state in polar media [18].
The theoretical framework for understanding CT transitions has evolved from Marcus theory, which classically describes electron transfer rates, to modern computational approaches including density functional theory (DFT) and time-dependent DFT (TD-DFT) [20]. These methods allow precise prediction of CT transition energies and intensities by calculating electronic structures and simulating UV-Vis spectra [21] [22].
Molecular Orbital Interactions in Charge-Transfer Transitions
The following diagram illustrates the fundamental orbital interactions in charge-transfer transitions:
This diagram illustrates the two primary CT mechanisms in metal complexes. In LMCT transitions, electrons move from ligand-centered molecular orbitals to metal-centered orbitals, typically occurring when ligands possess high-energy filled orbitals and metals have low-energy vacant orbitals. Conversely, MLCT transitions involve electron transfer from metal-centered orbitals to ligand-centered orbitals, commonly observed in metals with relatively high-energy d-electrons and ligands with low-energy π* orbitals [19] [16] [17].
Experimental Characterization Methods
Spectroscopic Techniques
Multiple spectroscopic methods are employed to characterize charge-transfer complexes and their transitions:
- UV-Vis Spectroscopy: The primary technique for identifying CT transitions through the appearance of new absorption bands that don't exist in the individual spectra of donor and acceptor components [18]. CT bands are typically broad and appear at wavelengths longer than 400 nm [18].
Benesi-Hildebrand Analysis: Used to determine the formation constant (KCTC) and molar extinction coefficient (εCTC) of 1:1 CT complexes by measuring absorbance at varying donor/acceptor concentrations [18]. The modified equation applied is:
(\frac{Ca Cd}{A} = \frac{1}{K{CTC}\varepsilon{CTC}} + \frac{Ca + Cd}{\varepsilon_{CTC}})
where (Ca) and (Cd) represent acceptor and donor concentrations, and A is the measured absorbance [18].
- Stoichiometry Determination: Multiple methods including Job's continuous variation, photometric titration, and conductometric titration establish the donor:acceptor ratio in CT complexes, typically confirming 1:1 stoichiometry [18].
Comprehensive Workflow for Charge-Transfer Complex Analysis
The experimental characterization of charge-transfer complexes follows a systematic workflow encompassing preparation, analysis, and application phases:
This comprehensive workflow illustrates the integrated experimental and computational approach required for thorough characterization of charge-transfer complexes. The process begins with sample preparation where donors and acceptors are combined in appropriate solvents, typically resulting in immediate color formation indicating CT complexation [18]. Solution analysis employs UV-Vis spectroscopy to identify characteristic CT bands, followed by stoichiometric determination using Job's method and stability constant calculation via Benesi-Hildebrand analysis [18]. Advanced characterization includes solid-state analysis through FTIR, NMR, and X-ray diffraction, complemented by computational studies using DFT and TD-DFT to predict electronic structures and validate experimental findings [21] [22]. Bioactivity assessment forms the final stage, evaluating potential pharmaceutical applications through DNA binding studies and antimicrobial assays [18].
Quantitative Data in Charge-Transfer Transitions
Spectroscopic Parameters of Characterized Charge-Transfer Complexes
Table 1: Experimental spectroscopic parameters of documented charge-transfer complexes
| Complex System | CT Band (nm) | Formation Constant (KCTC) L·molâ»Â¹ | Molar Extinction Coefficient (εCTC) L·molâ»Â¹Â·cmâ»Â¹ | Stoichiometry (D:A) |
|---|---|---|---|---|
| OPD-DDQ (in AN) [18] | 434, 542, 588 | 125.09 × 10² | 9.84 × 10² | 1:1 |
| OPD-DDQ (in MeOH) [18] | 463 | 56.50 × 10² | 8.12 × 10² | 1:1 |
| Hâ‚‚Oâ‚‚-TMP [21] | 247.3, 212.5 | - | 139, 97* | - |
| Hâ‚‚Oâ‚‚-NOR [21] | 280.2, 239.2 | - | 142, 358* | - |
| Hâ‚‚Oâ‚‚-CIP [21] | 254.5, 239.5 | - | 887, 365* | - |
| Hâ‚‚Oâ‚‚-OFL [21] | 285.2, 268.2, 254.5 | - | 177, 832, 226* | - |
| Hâ‚‚Oâ‚‚-SMR [21] | 241.3, 205.5, 193.6 | - | 289, 318, 169* | - |
*Oscillator strengths (f) × 10³ from TD-DFT calculations [21]
Frontier Molecular Orbital Energies in Charge-Transfer Complexes
Table 2: HOMO-LUMO energy gaps calculated for charge-transfer complexes
| Complex System | HOMO Energy (eV) | LUMO Energy (eV) | HOMO-LUMO Gap (eV) |
|---|---|---|---|
| Hâ‚‚Oâ‚‚-TMP [21] | -5.85 | -0.49 | 5.36 |
| Hâ‚‚Oâ‚‚-NOR [21] | -5.82 | -1.39 | 4.43 |
| Hâ‚‚Oâ‚‚-CIP [21] | -5.81 | -1.39 | 4.42 |
| Hâ‚‚Oâ‚‚-OFL [21] | -5.56 | -1.36 | 4.20 |
| Hâ‚‚Oâ‚‚-SMR [21] | -5.98 | -1.27 | 4.71 |
The HOMO-LUMO energy gap provides critical insights into the chemical reactivity and kinetic stability of charge-transfer complexes. Systems with smaller energy gaps, such as Hâ‚‚Oâ‚‚-OFL (4.20 eV), typically exhibit higher chemical reactivity compared to those with larger gaps like Hâ‚‚Oâ‚‚-TMP (5.36 eV) [21]. These quantum chemical parameters enable prediction of charge transfer propensity and complex stability.
Charge-Transfer in Metal Complexes
Transition Metal Complexes
In transition metal chemistry, CT transitions produce intense colors in coordination compounds. The key transitions include:
- d-d Transitions: These involve electronic excitation between metal-centered d-orbitals. They are Laporte-forbidden, resulting in weak absorption bands with low molar absorptivity (ε typically 10-100 L·molâ»Â¹Â·cmâ»Â¹) [17].
- Charge-Transfer Transitions: In contrast to d-d transitions, CT transitions are fully allowed and produce intense absorption bands with high molar absorptivity (ε typically >1,000 L·molâ»Â¹Â·cmâ»Â¹) [17]. The energy required for MLCT transitions is generally higher than for d-d transitions, but they occur more efficiently [17].
The electronic factors controlling CT transitions in metal complexes can be tuned through strategic ligand design. For ruthenium polypyridyl complexes, incorporating electron-donating "push" ligands and electron-withdrawing "pull" ligands can systematically alter MLCT transition energies [16]. Substitution at specific positions on bipyridine ligands (4,4' vs 5,5' vs 6,6') significantly impacts the π* orbital energy, enabling precise tuning of absorption maxima from 500 nm to 580 nm [16].
Lanthanide Complexes
For lanthanide ions, LMCT transitions are predominantly observed for easily reducible ions like Eu³⺠and Yb³⺠with easily oxidized ligands [16]. These transitions occur when ligands possess sufficiently high-energy occupied orbitals that can donate electrons to vacant f-orbitals on the metal center. The CT behavior in lanthanide complexes differs fundamentally from transition metals due to the core-like nature of 4f orbitals, resulting in distinctive photophysical properties.
Charge-Transfer in Drug Molecules and Pharmaceutical Applications
Antibiotic Charge-Transfer Complexes
Charge-transfer interactions play significant roles in pharmaceutical sciences, particularly in understanding drug-receptor interactions and antibiotic mechanisms. Studies on fluoroquinolone antibiotics (norfloxacin, ciprofloxacin, ofloxacin) with hydrogen peroxide demonstrate characteristic CT transitions between 239-285 nm, with oscillator strengths quantified through TD-DFT calculations [21]. These computational studies provide insights into the electronic structures responsible for antibiotic activity and potential degradation pathways.
The biological relevance of CT complexes extends to DNA binding interactions. Studies on the OPD-DDQ charge-transfer complex revealed intercalative binding with calf thymus DNA, exhibiting a substantial binding constant of 6.0 × 10âµ L·molâ»Â¹ [18]. This suggests potential biological implications for CT complexes in pharmaceutical contexts, possibly interfering with DNA replication processes in microorganisms.
Analytical and Bioactivity Applications
Pharmaceutical applications of charge-transfer complexes include:
- Bioactivity: CT complexes often exhibit enhanced antimicrobial properties compared to their individual components. The OPD-DDQ complex demonstrated notable antibacterial activity exceeding the standard drug tetracycline in some assessments [18].
- Drug-Receptor Studies: CT interactions serve as models for understanding drug-receptor binding mechanisms, as these complexes mimic the electron donor-acceptor interactions that frequently occur in biological systems [18].
- Analytical Detection: CT-based methods enable detection of non-chromophoric compounds that are otherwise difficult to analyze by conventional UV-Vis spectroscopy [23]. Metal complex-based CT interactions facilitate "switch on/off" sensing strategies for various analytes.
Computational Approaches and Theoretical Modeling
Density Functional Theory Applications
Modern computational methods, particularly density functional theory (DFT), have become indispensable tools for studying charge-transfer transitions:
- Geometry Optimization: DFT calculations at the B3LYP/6-31G(d,p) level provide optimized ground-state structures of CT complexes, identifying the most stable binding configurations between donors and acceptors [21] [22].
- TD-DFT Calculations: Time-dependent DFT predicts electronic excitation energies and oscillator strengths, allowing direct comparison with experimental UV-Vis spectra [21] [22]. These calculations successfully reproduce CT band positions and intensities.
- Molecular Orbital Analysis: HOMO-LUMO energy calculations quantify charge transfer propensity and chemical reactivity [21]. Electron density distributions reveal preferred sites for donor-acceptor interactions.
Marcus Theory and Electron Transfer Kinetics
The Marcus-Hush theory provides the fundamental framework for understanding electron transfer kinetics in charge-transfer processes [19] [20]. This classical theory relates the rate constant for electron transfer to the reorganization energy (λ) and the thermodynamic driving force (ΔG°):
[ k{ET} = \frac{2\pi}{\hbar} |H{AB}|^2 \frac{1}{\sqrt{4\pi\lambda kB T}} \exp\left[-\frac{(\lambda + \Delta G^\circ)^2}{4\lambda kB T}\right] ]
Where (H_{AB}) is the electronic coupling matrix element between donor and acceptor states. For outer-sphere electron transfer reactions, the Franck-Condon principle restricts electron transfer to nuclear configurations where the donor and acceptor states are degenerate, requiring vibrational excitation to achieve suitable geometry [19].
Essential Research Reagents and Materials
Table 3: Key research reagents and materials for charge-transfer studies
| Reagent/Material | Function in CT Research | Example Applications |
|---|---|---|
| Electron Acceptors | Component that receives electron density in CT complex | DDQ, chloranil, TCNQ [18] |
| Electron Donors | Component that donates electron density in CT complex | O-phenylenediamine, pharmaceuticals [18] |
| Polar Solvents | Medium for studying CT interactions in solution | Acetonitrile, methanol [18] |
| Deuterated Solvents | NMR characterization of CT complexes | DMSO-d6, CDCl3 [22] |
| Metal Salts | Synthesis of metal complexes for MLCT/LMCT studies | Cobalt(II) acetate, copper(II) acetate [22] |
| Schiff Base Ligands | Chelating ligands for stable metal complexes | Tetradentate Nâ‚‚Oâ‚‚ donors [22] |
| FT-IR Spectrophotometer | Structural characterization of CT complexes | KBr pellet method [22] |
| Computational Software | DFT/TD-DFT calculations of electronic structure | Gaussian 09, VASP [21] [22] |
Charge-transfer transitions represent a fundamental electronic process with significant implications across inorganic chemistry, pharmaceutical sciences, and materials research. The intense, solvent-sensitive nature of CT bands makes them readily identifiable in UV-Vis spectroscopy and provides valuable information about donor-acceptor interactions in molecular systems. For metal complexes, the distinction between LMCT and MLCT transitions depends critically on the relative orbital energies of metal centers and ligands, which can be systematically tuned through strategic ligand design. In pharmaceutical contexts, CT complexes model drug-receptor interactions and often exhibit enhanced bioactivity. Contemporary research continues to advance our understanding of these transitions through integrated experimental and computational approaches, particularly using TD-DFT to predict and rationalize CT band positions and intensities. The ongoing development of CT-based materials and pharmaceutical applications ensures continued relevance of these fundamental electronic transitions in spectroscopic research and drug development.
Electronic spectroscopy serves as a fundamental tool for probing the electronic structure of atoms and molecules, particularly in the development and characterization of pharmaceutical compounds. The intensity of spectral bands is governed by quantum mechanical selection rules that dictate the probability of electronic transitions. This whitepaper provides an in-depth examination of the two primary selection rules—the Spin Selection Rule and the Laporte Selection Rule—which define the allowedness or forbiddenness of transitions in UV-Vis spectroscopy. Framed within the context of electronic transition theory, this guide explores the theoretical foundations, practical implications, and exceptions to these rules, with specific application to transition metal complexes relevant to coordination chemistry and drug development research. The comprehensive analysis includes quantitative spectral data, experimental methodologies for observing these transitions, and visual tools to aid researchers in interpreting complex spectroscopic data.
In UV-Vis spectroscopy, the absorption of electromagnetic radiation promotes electrons from ground states to excited states, producing characteristic spectra that reveal critical information about molecular structure and energy levels. However, not all conceivable transitions between electronic states are equally probable; some transitions yield intense absorption bands, while others are faint or unobservable under standard conditions. Selection rules are quantum mechanical guidelines that predict which electronic transitions are "allowed" (high probability) and which are "forbidden" (low probability) based on symmetry and spin considerations [24]. These rules are paramount for researchers and drug development professionals who utilize spectroscopy to characterize metal complexes, organic chromophores, and pharmaceutical compounds.
The intensity of an absorption band is directly proportional to the transition probability, quantified by the molar absorptivity (ε). Forbidden transitions, while possible under certain conditions, exhibit significantly lower intensities than their allowed counterparts [24]. Two principal selection rules govern electronic transitions in centrosymmetric molecules and transition metal complexes: the Spin Selection Rule and the Laporte Selection Rule. Understanding their individual and combined effects is essential for accurate spectral interpretation and for designing compounds with desired photophysical properties in pharmaceutical applications.
The Laporte Selection Rule
Theoretical Foundation
The Laporte Selection Rule is a symmetry-based selection rule that applies rigorously to atoms and molecules possessing a center of inversion (centrosymmetric molecules) [25]. It states that electronic transitions that conserve parity (symmetry with respect to inversion) are forbidden. In practical terms, this means:
- Allowed transitions: Those involving a change in parity (
g → uoru → g) - Forbidden transitions: Those between states of the same parity (
g → goru → u) [24] [25]
The rule derives from the transition moment integral ∫ ψₑₗ μ̂ ψₑₗᵉˣ dτ, where ψₑₗ and ψₑₗᵉˣ are the wavefunctions of the initial and final electronic states, and μ̂ is the electric dipole moment operator [26]. This operator is odd under inversion. For the integral to be non-zero (indicating an allowed transition), the integrand must be even under inversion, which only occurs when the initial and final states have different parity [26].
In atomic orbitals, s and d orbitals are gerade (symmetric with respect to inversion), while p and f orbitals are ungerade (antisymmetric) [25]. Consequently, the Laporte Rule forbids pure s→s, p→p, d→d, and f→f transitions in centrosymmetric environments, but allows s→p, p→d, and d→f transitions, as these involve a change in parity [27].
Application to Transition Metal Complexes
The Laporte Rule is particularly significant in the electronic spectroscopy of transition metal complexes, where it explains the relative weakness of d-d transitions in octahedral complexes (which possess a center of inversion) [24] [25]. For example, in an octahedral complex, d orbitals have g symmetry, so transitions between them (g → g) are Laporte-forbidden [24]. Despite this forbiddenness, such transitions are observed in spectra but with low intensities (molar absorptivities generally below 100 L·molâ»Â¹Â·cmâ»Â¹), indicating that the selection rule can be relaxed under certain conditions [24] [25].
Table 1: Laporte Selection Rule Applications in Different Molecular Symmetries
| Molecular Geometry | Center of Inversion? | d-d Transition Status |
Typical Molar Absorptivity (ε, L·molâ»Â¹Â·cmâ»Â¹) |
|---|---|---|---|
| Octahedral (e.g., [Co(Hâ‚‚O)₆]²âº) | Yes | Laporte-forbidden | ~10 [25] |
| Tetrahedral (e.g., [CoClâ‚„]²â») | No | Laporte-allowed | ~600 [25] |
| Linear | Yes | Laporte-forbidden | Low (≤100) [24] |
| Square Planar | No | Laporte-allowed | Moderate to High |
Mechanisms of Rule Relaxation
Despite the theoretical forbiddenness, d-d transitions are observed in octahedral complexes due to two primary mechanisms that relax the Laporte Rule:
Vibronic Coupling: Molecular vibrations cause temporary distortions that disrupt the center of inversion. During these asymmetric vibrations, the
gandusymmetry labels become undefined, momentarily allowing the forbidden transition [24] [25]. Transitions occurring through this mechanism are called vibronic transitions. This is the dominant reason whyd-dbands are observable, albeit weak, in octahedral complexes [24].Orbital Mixing: In complexes with π-donor or π-acceptor ligands, metal
dorbitals can mix with ligand orbitals of different parity (e.g.,porbitals). This mixing introduces someucharacter into the predominantlygcharacterdorbitals, making the transitions partially allowed [24]. Additionally, in non-centrosymmetric geometries like tetrahedral complexes, the Laporte Rule does not apply at all, andd-dtransitions are allowed, resulting in more intense absorption bands [24] [25]. For instance, the tetrahedral [CoCl₄]²⻠complex (ε ≈ 600) has significantly more intense coloration than the octahedral [Co(H₂O)₆]²⺠complex (ε ≈ 10) [25].
Figure 1: The Laporte Rule and its relaxation mechanisms in centrosymmetric molecules. Although d-d transitions are formally forbidden, they become weakly observable through vibronic coupling and orbital mixing.
The Spin Selection Rule
Theoretical Foundation
The Spin Selection Rule dictates that electronic transitions must not involve a change in the total spin angular momentum of the system. Formally, this requires that ΔS = 0, meaning the spin multiplicity of the system remains unchanged [24] [28]. This rule arises because the electric dipole operator, which governs the interaction with radiation, does not act on electron spin. Therefore, a transition cannot directly flip the spin of an electron.
In the term symbols used to represent electronic states, the spin multiplicity is indicated by the left-hand superscript. The Spin Selection Rule thus allows transitions between states with the same superscript, such as ¹S → ¹P or ³T → ³A, but forbids transitions between states with different spin multiplicities, such as ³T → ¹D (a singlet-triplet transition) [24].
Practical Implications and Exceptions
Spin-forbidden transitions exhibit extremely low probabilities and consequently yield very faint spectral bands, with molar absorptivities (ε) often in the range of 10â»Â³ to 1 L·molâ»Â¹Â·cmâ»Â¹ [27]. A classic example is the octahedral complex [Mn(Hâ‚‚O)₆]²âº; its manganese(II) ion has a dâµ high-spin configuration. The ground state is â¶Aâ‚g, and the lowest-energy excited states are quartet states (â´Tâ‚g, â´Tâ‚‚g). Transitions to these states are both Laporte-forbidden (being g→g) and spin-forbidden (ΔS = 1). The combined effect of these prohibitions results in such weak absorption that dilute solutions of this complex appear nearly colorless [27] [28].
The primary mechanism for relaxation of the Spin Selection Rule is spin-orbit coupling. This is an electromagnetic interaction between the electron's spin and its orbital motion around the nucleus. It effectively mixes pure spin states, creating new wavefunctions that are admixtures of different spin multiplicities (e.g., mixing some singlet character into a triplet state) [28]. This mixing provides a pathway for otherwise forbidden transitions to gain a small but non-zero probability. The strength of spin-orbit coupling increases with the atomic number of the element, making spin-forbidden transitions more pronounced for heavier metal ions like those in the second and third transition series, as well as lanthanides and actinides [25].
Combined Effect of Selection Rules and Experimental Manifestations
Quantitative Spectral Intensities
In transition metal complexes, the Spin and Laporte selection rules often operate simultaneously. The observed intensity of an absorption band depends on the degree to which each rule is obeyed or violated. The table below categorizes transition types based on their adherence to these rules and their resulting spectral intensities.
Table 2: Classification of Electronic Transitions by Selection Rules and Resulting Intensities
| Transition Type | Spin Rule | Laporte Rule | Typical εmax (L·molâ»Â¹Â·cmâ»Â¹) | Example Complex |
|---|---|---|---|---|
| Spin & Laporte Forbidden | Violated | Violated | 10â»Â³ - 1 | [Mn(Hâ‚‚O)₆]²⺠[27] |
| Spin Allowed, Laporte Forbidden | Obeyed (ΔS=0) | Violated | 1 - 100 | [Cr(H₂O)₆]³⺠[24] [27] |
| Spin Allowed, Laporte Allowed | Obeyed (ΔS=0) | Obeyed (e.g., g→u) | 100 - 1000 | Tetrahedral [CoCl₄]²⻠[25] [27] |
| Charge-Transfer | Often Obeyed | Often Obeyed | 1,000 - 10ⶠ| [MnOâ‚„]â», [Fe(CN)₆]³⻠[27] |
Experimental Protocols for Electronic Spectroscopy
The following section outlines a generalized experimental methodology for acquiring and interpreting UV-Vis-NIR spectra of transition metal complexes to assess the operation of selection rules.
Sample Preparation
- Reagent Solutions: Prepare a solution of the complex under study at a concentration typically between 1×10â»âµ M and 1×10â»Â² M, depending on the expected absorption intensity. For weakly absorbing (highly forbidden) transitions, higher concentrations are required.
- Solvent Selection: Choose a solvent that is transparent in the spectral region of interest and does not coordinate with the metal ion in a way that alters the complex's geometry. Common choices include water, acetonitrile, and dichloromethane.
- Cuvette Selection: Use spectrophotometric cuvettes with appropriate path lengths (usually 1 cm) and material (e.g., quartz for UV-Vis, silica or glass for visible region only).
Data Acquisition
- Baseline Correction: Collect a baseline spectrum using a cuvette filled only with the pure solvent.
- Sample Measurement: Obtain the absorption spectrum of the complex solution across the relevant wavelength range (e.g., 200-800 nm for UV-Vis, or extending to ~2500 nm for NIR if needed for certain
d-dorf-ftransitions). - Parameter Settings: Use a moderate spectral bandwidth (e.g., 1-2 nm) to resolve individual bands without excessive loss of light intensity. Perform multiple scans and average them to improve the signal-to-noise ratio, which is crucial for detecting weak forbidden bands.
Data Analysis and Interpretation
- Band Assignment: Identify the number, position (energy, λmax in nm or cmâ»Â¹), and intensity (εmax) of absorption bands.
- Intensity as a Diagnostic Tool:
- Weak bands (ε < 100): Suggest either spin-forbiddenness, Laporte-forbiddenness, or both. This is characteristic of
d-dtransitions in centrosymmetric complexes. - Moderate bands (ε ~ 10²-10³): Often indicate Laporte-allowed transitions, such as
d-dtransitions in tetrahedral complexes or those involving significant orbital mixing. - Intense bands (ε > 10³): Typically signify fully allowed transitions, most commonly Charge-Transfer (CT) transitions (e.g., Ligand-to-Metal or Metal-to-Ligand Charge Transfer) [27].
- Weak bands (ε < 100): Suggest either spin-forbiddenness, Laporte-forbiddenness, or both. This is characteristic of
- Comparison with Theoretical Predictions: Compare the observed spectrum with energy level diagrams (e.g., Tanabe-Sugano diagrams) and selection rule predictions to assign specific electronic transitions (e.g.,
â´Aâ‚‚g → â´Tâ‚‚gfor[Cr(Hâ‚‚O)₆]³âº).
Figure 2: A workflow for diagnosing the nature of an electronic transition based on its molar absorptivity (ε_max), using the selection rules as a guide.
The Scientist's Toolkit: Research Reagents and Materials
Table 3: Essential Materials and Reagents for Studying Electronic Transitions
| Item | Function/Application |
|---|---|
Transition Metal Salts (e.g., CoCl₂, KMnO₄, Cr(NO₃)₃) |
Source of metal centers for synthesizing coordination complexes with varied d-electron configurations. |
Spectroscopic Grade Solvents (e.g., H₂O, CH₃CN, CH₂Cl₂) |
Dissolve complexes without introducing significant background absorption in the UV-Vis region. |
Ligand Library (e.g., Hâ‚‚O, NH₃, CNâ», Clâ», phenanthroline, porphyrins) |
Create complexes with different geometries (octahedral, tetrahedral), ligand field strengths, and electronic properties to probe selection rules. |
| Quartz Cuvettes (path lengths 1 cm, 0.1 cm, etc.) | Hold liquid samples for measurement; quartz is transparent from UV to IR. |
| UV-Vis-NIR Spectrophotometer | Instrument for measuring absorption spectra across the ultraviolet, visible, and near-infrared regions. |
| Tanabe-Sugano Diagrams | Theoretical charts for correlating observed transition energies with ligand field parameters (Δ_o, B) in d⿠metal complexes. |
| RGT-018 | RGT-018, MF:C27H24F3N7O2, MW:535.5 g/mol |
| APY29 | APY29, MF:C17H16N8, MW:332.4 g/mol |
The Spin and Laporte selection rules provide the fundamental theoretical framework for understanding the intensities of bands in electronic spectra. While the Spin Selection Rule (ΔS = 0) forbids transitions that change the total spin, the Laporte Rule forbids transitions that conserve parity in centrosymmetric molecules. Forbidden transitions, a hallmark of d-d spectra in octahedral complexes, are observed with low intensity due to relaxation mechanisms like vibronic coupling, orbital mixing, and spin-orbit coupling.
For researchers in drug development and materials science, these rules are not mere theoretical constructs but essential tools. They enable the decoding of complex spectra to extract information on oxidation states, coordination geometry, and ligand environment of metal centers in biologically relevant complexes and catalysts. The ability to distinguish between Laporte-forbidden d-d transitions, fully allowed charge-transfer bands, and the transitions of organic chromophores is critical for assigning spectral features and designing molecules with tailored photophysical and photocatalytic properties. Mastery of these governing rules empowers scientists to fully leverage UV-Vis spectroscopy as a powerful probe of electronic structure.
Within the foundational theory of electronic transitions underpinning UV-Vis spectroscopy, the Franck-Condon Principle stands as a cornerstone for interpreting spectral profiles. This principle asserts that electronic transitions occur on a time scale vastly shorter than nuclear motion, resulting in vertical transitions that simultaneously populate vibrational states in the excited electronic manifold. The probability of these vibronic transitions is governed by the overlap of vibrational wavefunctions, quantitatively expressed by the Franck-Condon factor. This whitepaper provides an in-depth examination of the principle's quantum mechanical foundation, its manifestation in absorption and emission spectra, and detailed methodologies for its application in quantitative spectroscopic analysis, with particular relevance for researchers in molecular spectroscopy and drug development.
The interaction of light with matter, particularly the absorption of ultraviolet or visible light by molecules, excites electrons from their ground state to higher energy levels. However, a comprehensive understanding of UV-Vis spectra extends beyond purely electronic transitions. Within any electronic state, a molecule exists in a set of quantized vibrational energy levels. The total energy of a molecule is thus the sum of its electronic, vibrational, and rotational energies. While rotational fine structure is often unresolved in solution-phase spectra of complex molecules, the vibrational progression remains a critical feature shaping the absorption band profile.
The Franck-Condon Principle provides the theoretical framework for understanding the intensities of these vibronic transitions—the coupled electronic and vibrational transitions. Formulated by James Franck and Edward Condon in 1926, the principle posits that "an electronic transition takes place so rapidly that a vibrating molecule does not change its internuclear distance appreciably during the transition" [29]. This is a direct consequence of the Born-Oppenheimer approximation, which allows for the separation of electronic and nuclear wavefunctions due to the significant mass disparity between electrons and nuclei [30] [31]. During the femtosecond-scale electronic transition, the nuclei are effectively frozen in place, leading to a vertical transition on a potential energy diagram [32].
Theoretical Foundation of the Franck-Condon Principle
Quantum Mechanical Formulation
The quantum mechanical treatment of the Franck-Condon principle derives from the transition probability, ( P ), between an initial state ( |\psi{initial}\rangle ) and a final state ( |\psi{final}\rangle ), which is proportional to the square of the transition dipole moment matrix element [31]:
[ P{i \rightarrow f} = \left| \langle \psi^{*}{final} | \boldsymbol{\mu} | \psi_{initial} \rangle \right|^2 ]
The total wavefunction can be separated into electronic (( \psi{el} )), vibrational (( \psi{v} )), and spin (( \psi{s} )) components: ( \psi{total} = \psi{el} \psi{v} \psi_{s} ) [30]. Within the Condon approximation, which assumes the electronic transition moment is independent of nuclear coordinates, the transition probability simplifies to a product of factors:
[ P \propto \underbrace{ \left| \int \psi{v}'^{*} \psi{v} d\tau{n} \right|^2 }{\text{Franck-Condon Factor}} \times \underbrace{ \left| \int \psi{e}'^{*} \mu{e} \psi{e} d\tau{e} \right|^2 }{\text{Orbital Selection Rule}} \times \underbrace{ \left| \int \psi{s}'^{*} \psi{s} d\tau{s} \right|^2 }_{\text{Spin Selection Rule}} ]
The first term, the square of the overlap integral of the initial and final vibrational wavefunctions, is the Franck-Condon Factor. It is this factor that dictates the relative intensity of vibrational transitions within an electronic absorption or emission band [30] [31]. The subsequent terms enforce the orbital and spin selection rules for the electronic transition itself.
The Semiclassical Picture: Vertical Transitions
The semiclassical interpretation provides an intuitive picture: because the electronic transition is instantaneous relative to nuclear vibration, it is represented by a vertical arrow on a potential energy curve (or surface) diagram, where the x-axis represents nuclear coordinates (e.g., internuclear distance in a diatomic molecule) and the y-axis represents the potential energy [30] [32]. The most probable transition originates from the most probable nuclear configuration of the initial state. For a molecule in the vibrational ground state (v=0), this is typically at the classical turning points of the oscillation. The vertical arrow projects this configuration upward to the potential energy curve of the excited electronic state. The vibrational level in the excited state that is intersected by this vertical line has the greatest spatial overlap with the initial vibrational wavefunction and thus the highest transition probability [30] [29].
Manifestations in Absorption Spectroscopy
The shape of an electronic absorption band is a direct fingerprint of the vibrational level structure in the excited state and the shift in equilibrium geometry between the two electronic states.
Case Studies in Band Profile Analysis
The interplay between the potential energy surfaces of the ground and excited states leads to distinct spectral profiles, summarized in the table below.
Table 1: Franck-Condon Principle Case Studies and Resulting Spectral Profiles
| Case | Equilibrium Geometry Shift | Most Probable Transition | Spectral Profile | Example Molecule |
|---|---|---|---|---|
| Case 1 | Minimal or No Shift | v"=0 → v'=0 | Strong 0-0 band; rapidly diminishing intensity for higher v' [29] | O₂ [29] |
| Case 2 | Small Shift | v"=0 → v'=2 (example) | Intensity maximum at a non-zero v' band; structured progression [29] | CO [29] |
| Case 3 | Large Shift | v"=0 → high v' (near dissociation) | Continuum spectrum with no vibrational structure [29] | I₂ [29] |
The Franck-Condon Factor and Absorption Intensity
The intensity of an absorption peak corresponding to a transition from the ground vibrational level of the electronic ground state (v") to a specific vibrational level of the electronic excited state (v') is proportional to the Franck-Condon factor, ( \left| \langle \psi{v'} | \psi{v"} \rangle \right|^2 ) [30] [31]. This factor quantifies the degree of spatial overlap between the two vibrational wavefunctions. When the potential energy curves are aligned, the 0-0 transition has the largest overlap. A shift in the equilibrium geometry reduces the overlap for the 0-0 transition and increases it for transitions to higher vibrational levels (v' > 0), as illustrated in the diagram below.
Experimental Protocols and Data Interpretation
Protocol for Measuring and Assigning Vibronic Structure
Objective: To record the UV-Vis absorption spectrum of a molecule with vibrational resolution and assign the vibronic progression using Franck-Condon analysis.
Materials and Reagents:
- High-purity analyte molecule (e.g., organic dye, polyatomic aromatic hydrocarbon).
- Spectrophotometric-grade solvent (e.g., n-hexane, cyclohexane).
- Quartz cuvettes (path length 1 cm).
- High-resolution UV-Vis/NIR spectrophotometer.
- Thermostatted cell holder.
- Data analysis software (e.g., Python with NumPy/SciPy, MATLAB, Origin).
Methodology:
- Sample Preparation: Prepare a dilute solution of the analyte (typical absorbance < 1.0 at the peak maximum) to minimize aggregation and inner-filter effects. Degas the solution if necessary to eliminate oxygen quenching.
- Instrumental Configuration: Use a spectrophotometer capable of a spectral bandwidth less than 0.5 nm. Maintain a constant, low temperature (e.g., 77 K using a cryostat or 100-150 K in a glass-forming solvent) to reduce thermal broadening of vibrational lines [30]. Scan at a slow speed with high data density.
- Data Collection: Record the baseline-corrected absorption spectrum across the relevant electronic transition.
- Peak Assignment: Identify the peak with the shortest wavelength/largest energy, which is assigned as the 0-0 transition [30]. Subsequent peaks at lower energies are assigned as 0-1, 0-2, etc., corresponding to transitions to successively higher vibrational levels (v'=1, v'=2, ...) in the excited electronic state.
- Franck-Condon Analysis:
- Measure the relative areas under each vibronic peak to obtain experimental intensities.
- Construct a harmonic (or anharmonic) oscillator model for the ground and excited states.
- Calculate the Franck-Condon factors for the various v"=0 to v' transitions.
- Iteratively adjust the model parameters (e.g., the shift in equilibrium position, vibrational frequencies) until the calculated Franck-Condon factors match the experimentally observed intensity pattern.
The Researcher's Toolkit: Essential Reagents and Materials
Table 2: Key Research Reagent Solutions and Materials for Franck-Condon Studies
| Item | Function & Importance |
|---|---|
| Spectrophotometric-Grade Solvents | High-purity solvents with low UV-Vis cutoff are essential to avoid spurious absorption bands that obscure the vibrational structure of the analyte. |
| Quzrtz Cuvettes | Required for UV-Vis transmission measurements. Matched cuvettes for sample and reference ensure accurate baseline correction. |
| Cryostat or Cooling Cell Holder | Cooling the sample reduces thermal energy, which narrows the individual vibrational linewidths, revealing resolved vibronic structure [30]. |
| High-Resolution Spectrophotometer | Instrumentation with fine spectral bandwidth and high signal-to-noise ratio is critical for resolving closely spaced vibrational peaks. |
| Computational Chemistry Software | Software for calculating molecular orbitals, potential energy surfaces, and vibrational wavefunctions is used for quantitative Franck-Condon simulation and fitting. |
| Ripa-56 | Ripa-56, MF:C13H19NO2, MW:221.29 g/mol |
| PF-06928215 | PF-06928215, MF:C20H20N4O4, MW:380.4 g/mol |
Advanced Applications and Contemporary Relevance
The Franck-Condon principle remains a vital tool in modern chemical physics and materials science. Its application has been extended beyond traditional spectroscopy to domains like mechanochemistry [33]. In the "sudden-force" regime, where an external force is applied rapidly to a molecule, the transition to a force-modified potential energy surface is analogous to an optical electronic transition. This can create a non-equilibrium, vibrationally excited ensemble of molecules, providing an additional activation source beyond simple barrier reduction, and leading to non-statistical reaction kinetics [33].
Furthermore, the principle is foundational for understanding electron transfer reactions in condensed phases, as described by Marcus Theory [34]. These reactions are governed by the Franck-Condon principle that electron transfer occurs at fixed nuclear positions, requiring the reorganization of the nuclear framework (solvent and molecular skeleton) to a configuration where the initial and final states are isoenergetic before the electron can tunnel.
The Franck-Condon Principle provides the essential link between the quantum mechanics of molecular structure and the observed profiles of electronic absorption bands. By quantifying the probabilities of vibronic transitions through wavefunction overlap, it allows researchers to decode geometric changes upon electronic excitation from experimental spectra. The principle's utility spans from interpreting the structured bands of small molecules in the gas phase to understanding the broad, yet asymmetric, bands of chromophores in solution and solid states. For researchers in drug development, where UV-Vis spectroscopy is routinely used for compound identification and quantification, a deeper understanding of the Franck-Condon principle offers insights into the electronic and vibrational characteristics of chromophores, which can inform the design of molecules with tailored photophysical properties. Mastery of this core principle is therefore indispensable for any scientist leveraging electronic spectroscopy to probe molecular identity, structure, and dynamics.
From Spectrum to Structure: A Step-by-Step Guide to Interpreting UV-Vis Data
Ultraviolet-Visible (UV-Vis) spectroscopy is a fundamental analytical technique that probes the electronic structure of molecules by measuring their interaction with ultraviolet and visible light [11]. This interaction involves the promotion of electrons from ground state orbitals to higher-energy excited states, a process known as an electronic transition [4]. The energy required for these transitions corresponds to specific wavelengths of light in the UV-Vis range, typically between 200-800 nm [35] [11].
When a molecule absorbs light, electrons are excited from the highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO) [4]. The energy difference between these orbitals (ΔE) determines the wavelength of light absorbed, following the relationship ΔE = hν, where h is Planck's constant and ν is the frequency of the absorbed light [11] [4]. This foundational principle enables researchers to extract critical information about molecular structure, concentration, and electronic properties through two key parameters: lambda max (λmax) and molar absorptivity (ε) [36] [37].
Lambda Max (λmax): The Signature of Electronic Transitions
Fundamental Definition and Significance
Lambda max (λmax) is the specific wavelength at which a substance exhibits maximum absorption of light [35]. This parameter serves as a characteristic fingerprint for molecules and chromophores (light-absorbing groups), providing crucial insights into their electronic structure [36]. The position of λmax reveals the energy gap between ground and excited states, with longer wavelengths indicating smaller energy gaps required for electronic transitions [4].
The value of λmax is intrinsically linked to molecular structure. For example, in ethene (which contains a single carbon-carbon double bond), the π→π* transition occurs at approximately 171-174 nm [36] [4]. As conjugation increases, the energy gap between HOMO and LUMO decreases, resulting in a bathochromic shift (red shift) of λmax to longer wavelengths [4]. This phenomenon explains why extended conjugated systems often absorb in the visible region, appearing colored to the human eye [4].
Structural Factors Influencing Lambda Max
The position of λmax is profoundly influenced by molecular structure, particularly the presence and extent of conjugation [4]. The following table summarizes how structural features affect λmax:
Table 1: Structural Factors Influencing Lambda Max (λmax)
| Structural Feature | Effect on λmax | Example/Mechanism |
|---|---|---|
| Isolated π bond | Lower λmax | Ethene: ~174 nm [4] |
| Conjugated π system | Increases λmax | Buta-1,3-diene: 217 nm [36] |
| Extended conjugation | Further increases λmax | β-carotene: ~450 nm (visible region) [11] [4] |
| Auxochromes | Varies by substituent | -OH, -NHâ‚‚ can cause bathochromic shifts [35] |
| Exocyclic double bond | +5 nm per bond | Double bond extending outside a ring system [35] |
The underlying molecular orbital theory explains these trends: conjugation creates a system of overlapping p-orbitals that delocalize π electrons, narrowing the HOMO-LUMO gap and reducing the energy required for electronic transitions [4]. This relationship between structure and λmax enables researchers to make predictions about molecular characteristics based on spectroscopic data.
Molar Absorptivity (ε): Quantifying Transition Probability
Fundamental Definition and Significance
Molar absorptivity (ε), also known as the molar extinction coefficient, is a measure of how strongly a chemical species absorbs light at a particular wavelength [38] [37]. This parameter is intrinsically related to the probability of an electronic transition occurring, with higher ε values indicating more allowed transitions [11]. Mathematically, ε is defined by the Beer-Lambert law as the proportionality constant between absorbance and the product of concentration and path length [38].
Molar absorptivity values span several orders of magnitude, providing insights into the nature of electronic transitions [35]:
- Strongly absorbing compounds: ε > 10,000 L·molâ»Â¹Â·cmâ»Â¹
- Weakly absorbing compounds: ε = 10-100 L·molâ»Â¹Â·cmâ»Â¹
These differences reflect fundamental selection rules governing electronic transitions. For example, the weak absorption band of benzene at ~260 nm (ε ≈ 200) is attributed to a symmetry-forbidden π→π* transition, while the intense absorption of β-carotene at ~450 nm represents a fully allowed π→π* transition [11].
Transition Types and Their Characteristics
Different electronic transitions exhibit characteristic ε values, providing information about their probability and the molecular orbitals involved [11]:
Table 2: Electronic Transitions and Typical Molar Absorptivity Values
| Transition Type | Typical ε Value (L·molâ»Â¹Â·cmâ»Â¹) | Characteristics | Example |
|---|---|---|---|
| π → π* | 10,000-100,000 | High probability, allowed | Conjugated dienes, aromatic compounds [11] |
| n → π* | 10-100 | Low probability, forbidden | Carbonyl compounds [11] |
| σ → σ* | 1,000-10,000 | Very high energy, deep UV | Alkanes, molecular hydrogen [4] |
| n → σ* | 1,000-10,000 | Medium energy | Alcohols, amines [11] |
| Charge-transfer | 10,000-100,000 | Very intense, broad bands | Metal complexes, donor-acceptor systems [11] |
The intensity of an electronic transition is proportional to the square of the transition dipole moment, with allowed transitions having larger transition dipole moments and thus higher intensities [11]. Forbidden transitions can still occur with lower intensity due to vibronic coupling, which relaxes the selection rules by mixing vibrational and electronic wavefunctions [11].
The Beer-Lambert Law: The Fundamental Relationship
Mathematical Formulation and Components
The Beer-Lambert Law establishes the fundamental relationship between light absorption and the properties of the absorbing species, providing the mathematical foundation for quantitative analysis using UV-Vis spectroscopy [39] [38]. The law is expressed as:
A = ε · c · l
Where:
- A is the absorbance (unitless)
- ε is the molar absorptivity (L·molâ»Â¹Â·cmâ»Â¹)
- c is the molar concentration (mol·Lâ»Â¹)
- l is the path length (cm) [38] [40]
Absorbance is defined logarithmically in terms of light intensities: A = logâ‚â‚€(Iâ‚€/I), where Iâ‚€ is the incident light intensity and I is the transmitted light intensity [39] [38]. This logarithmic relationship means that an absorbance of 1 corresponds to 10% transmittance, while an absorbance of 2 corresponds to 1% transmittance [39].
Practical Applications and Limitations
The Beer-Lambert Law enables the determination of solute concentrations in solutions by measuring absorbance [36] [39]. For example, if the molar absorptivity of a compound is known, its concentration can be calculated from a single absorbance measurement [36]. However, for highest accuracy, the preferred method involves constructing a calibration curve by measuring absorbance values of several standard solutions of known concentration [36] [39].
The linear relationship between absorbance and concentration holds true for dilute solutions, but deviations can occur at higher concentrations due to factors such as molecular interactions and instrumental limitations [36]. The following diagram illustrates the experimental workflow for quantitative analysis using the Beer-Lambert Law:
Diagram 1: Workflow for quantitative analysis using UV-Vis spectroscopy and the Beer-Lambert Law
Experimental Protocols and Methodologies
Instrumentation and Measurement Techniques
UV-Vis spectroscopy employs a spectrophotometer that typically consists of several key components [35] [40]:
- Light source: Provides broad-spectrum UV and visible light
- Monochromator: Separates light into specific wavelengths using prism or diffraction grating
- Sample compartment: Holds cuvettes containing sample and reference solutions
- Detector: Measures the intensity of transmitted light
The fundamental measurement involves comparing the light intensity passing through a sample solution (I) to that passing through a reference blank (Iâ‚€) containing only solvent [35]. Modern instruments automatically scan across wavelengths and compute absorbance values, generating a plot of absorbance versus wavelength known as an absorption spectrum [35].
For accurate results, several experimental factors must be controlled:
- Solvent selection: Must be transparent in the spectral region of interest [35]
- Cuvette path length: Typically 1 cm, must be known precisely [36]
- Concentration range: Must ensure absorbance values remain within the linear range (generally A < 1-2) [36] [38]
- Wavelength selection: Typically performed at λmax for maximum sensitivity [36] [40]
Quantitative Analysis Protocol
The following step-by-step protocol describes the determination of compound concentration using UV-Vis spectroscopy:
Sample Preparation:
- Prepare a stock solution of the analyte with accurately known concentration
- Create a series of standard solutions by dilution to bracket the expected unknown concentration
- Ensure the solvent is optically transparent in the spectral region of interest [35]
Spectrum Acquisition:
Calibration Curve Construction:
Unknown Sample Analysis:
This methodology is illustrated by the example of determining Red-40 concentration in Gatorade, where an absorbance of 0.500 with ε = 2.13×10â´ L·molâ»Â¹Â·cmâ»Â¹ and path length of 1.0 cm yielded a concentration of 2.3×10â»âµ mol/L [40].
The Researcher's Toolkit: Essential Materials and Reagents
Successful implementation of UV-Vis spectroscopy requires specific materials and reagents, each serving distinct functions in the experimental process:
Table 3: Essential Research Reagents and Materials for UV-Vis Spectroscopy
| Item | Function/Purpose | Key Considerations |
|---|---|---|
| Spectrophotometer | Measures light absorption as a function of wavelength | Requires appropriate spectral range (typically 200-800 nm) [35] [37] |
| Cuvettes | Holds liquid samples for measurement | Must be optically transparent; quartz for UV, glass/plastic for visible [40] |
| Reference Solvents | Serves as blank and sample solvent | Must be optically pure and transparent in spectral region of interest [35] |
| Standard Compounds | Enables calibration and verification | High purity compounds with known ε values [36] |
| Volumetric Glassware | Prepares accurate solution concentrations | Precision volumetric flasks and pipettes for standard preparation [36] |
| MC-GGFG-Exatecan | MC-GGFG-Exatecan, CAS:1600418-29-8, MF:C49H51FN8O11, MW:947.0 g/mol | Chemical Reagent |
| BMVC | BMVC, CAS:627810-06-4, MF:C28H25I2N3, MW:657.3 g/mol | Chemical Reagent |
Advanced Applications in Research and Drug Development
UV-Vis spectroscopy finds diverse applications across chemical, biological, and materials research, particularly leveraging the information contained in λmax and ε values.
In drug development, UV-Vis spectroscopy provides critical information about molecular structure and concentration [37]. The technique can identify functional groups, detect conjugation patterns, and quantify drug compounds in solutions [36] [37]. The high sensitivity of modern instruments enables detection of very low concentrations (10â»âµ M or lower), making it valuable for analyzing precious compounds available only in small quantities [36].
Operando UV-Vis spectroelectrochemistry represents an advanced application where spectroscopy is combined with electrochemistry to study catalytic mechanisms under actual operating conditions [41]. This approach allows researchers to quantify the accumulation of reactive species at catalyst-electrolyte interfaces and characterize the kinetics of rate-determining steps in catalysis [41].
The relationship between molecular structure and spectral characteristics enables researchers to make predictions about λmax using empirical rules, such as Woodward-Fieser rules for conjugated dienes and Fieser-Kuhn rules for polyenes [35]. These tools facilitate structural elucidation and compound identification in complex mixtures.
Lambda max and molar absorptivity represent fundamental parameters in UV-Vis spectroscopy that provide deep insights into molecular electronic structure and enable precise quantitative analysis. The position and intensity of absorption bands serve as signatures of specific chromophores and electronic transitions, while the Beer-Lambert Law provides the mathematical foundation for concentration determinations. As research techniques continue to evolve, particularly through integration with other analytical methods, UV-Vis spectroscopy remains an indispensable tool for researchers across chemistry, materials science, and drug development.
Identifying Chromophores and Functional Groups in Bioactive Molecules
Ultraviolet-visible (UV-Vis) spectroscopy serves as a fundamental analytical technique for identifying chromophores and functional groups in bioactive molecules by measuring their interaction with light across the 100 to 900 nm wavelength range [42]. This interaction manifests as electronic transitions, where molecules absorb electromagnetic radiation, promoting electrons from ground state orbitals to higher energy excited states [6] [2]. The resulting absorption spectrum provides a characteristic fingerprint that reveals vital information about molecular structure, concentration, and electronic environment [42] [43].
The interpretation of these spectra is particularly valuable in pharmaceutical and biochemical research, where understanding the electronic properties of bioactive compounds informs drug design, development, and analysis [43]. The technique's non-destructive nature, cost-effectiveness, and rapid analysis capabilities make it suitable for various applications, from characterizing organic compounds and some inorganic species to studying polymer nanocomposites and metalloprotein complexes [43].
Theoretical Foundations of Electronic Transitions
Molecular Orbitals and Chromophores
At the core of UV-Vis spectroscopy lies the concept of molecular orbitals, which represent the probability distribution of electrons in a molecule [6]. When light energy matches the energy difference between molecular orbitals, electrons can undergo promotion from bonding orbitals (lower energy) to antibonding orbitals (higher energy) [5]. The Highest Occupied Molecular Orbital (HOMO) and the Lowest Unoccupied Molecular Orbital (LUMO) represent the frontier orbitals involved in these electronic transitions, with the energy gap between them determining the wavelength of absorbed light [6].
Chromophores constitute the light-absorbing molecular components responsible for color and UV absorption characteristics [44]. These specific functional groups contain valence electrons of relatively low excitation energy, enabling them to absorb light in the UV-visible region [5]. Common chromophores in bioactive molecules include C=C, C=O, aromatic rings, and conjugated systems [6].
Types of Electronic Transitions
Several types of electronic transitions can occur when molecules absorb UV-visible radiation, each with characteristic energy requirements and probabilities [45] [2]:
- π→π* transitions occur in molecules with unsaturation (e.g., alkenes, alkynes, aromatics) and typically exhibit high molar absorptivities (ε = 2,400-25,000 L·molâ»Â¹Â·cmâ»Â¹) due to good orbital overlap [45] [2]. For isolated double bonds, these transitions generally appear at shorter wavelengths (160-280 nm), but conjugation significantly shifts them to longer wavelengths [45].
- n→π* transitions involve the promotion of a non-bonding electron (typically on heteroatoms like O, N, S) to a Ï€* antibonding orbital [45]. These transitions are of lower energy (λmax = 279-320 nm) but have weak intensity (ε = 10-50 L·molâ»Â¹Â·cmâ»Â¹) due to poor orbital overlap [45] [2]. They are characteristic of carbonyl compounds, azo groups, and nitro compounds [44].
- n→σ* transitions occur in saturated compounds containing heteroatoms with lone pairs (e.g., alcohols, amines, thiols) and typically appear in the 160-250 nm range with moderate intensity (ε = 100-7,000 L·molâ»Â¹Â·cmâ»Â¹) [45] [2].
- σ→σ* transitions require the highest energy and appear at very short wavelengths (below 200 nm), making them less commonly observed in routine UV-Vis spectroscopy [2].
The following diagram illustrates the orbital interactions and relative energy requirements for these electronic transitions:
The Role of Conjugation
Conjugation, the alternating pattern of single and multiple bonds, dramatically influences the absorption characteristics of chromophores by extending electron delocalization across the molecular framework [44] [5]. This delocalization lowers the energy gap between the HOMO and LUMO orbitals, resulting in a bathochromic shift (red shift) where absorption moves to longer wavelengths [6]. Extended conjugation systems also typically exhibit hyperchromic effects (increased absorption intensity) due to enhanced transition probabilities [42].
The impact of conjugation is evident in natural chromophores such as chlorophyll, which possesses an extensively conjugated porphyrin ring system that absorbs strongly in both blue (~430 nm) and red (~660 nm) regions of the visible spectrum [44]. Similarly, synthetic dyes and pharmaceutical compounds often incorporate conjugated systems to tune their light absorption properties for specific applications [44].
Chromophores and Functional Groups in Bioactive Molecules
Characteristic Chromophores and Their Transitions
Bioactive molecules contain specific chromophores that dictate their UV-Vis absorption characteristics. The table below summarizes common chromophores found in pharmaceutical compounds and their typical absorption properties:
| Chromophore | Structure | Transition Type | Typical λmax (nm) | Molar Absorptivity (ε, L·molâ»Â¹Â·cmâ»Â¹) |
|---|---|---|---|---|
| Carbonyl | C=O | n→π* | 270-300 | 10-100 [42] [45] |
| Aromatic | Benzene ring | π→π* | 250-280 | 2,400-25,000 [42] [45] |
| Conjugated diene | C=C-C=C | π→π* | 220-250 | 10,000-25,000 [42] |
| Nitro group | NO₂ | n→π* | 350-400 (weak) | 10-50 [42] |
| π→π* | 200-250 (strong) | 5,000-10,000 [42] | ||
| Azo group | N=N | n→π* | 400-500 | Varies with conjugation [44] |
| Amide | CONH₂ | n→π* | 200-220 | ~100 [44] |
Auxochromes and Their Effects
Auxochromes are functional groups that themselves do not absorb strongly in the UV-Vis region but can modify the absorption properties of chromophores when attached to them [44]. These groups typically contain heteroatoms with non-bonded electron pairs that can interact with the chromophore's π-system [6]. Common auxochromes include hydroxyl (-OH), amino (-NH₂, -NHR, -NR₂), and thiol (-SH) groups [44].
Auxochromes can induce several types of spectral modifications:
- Bathochromic shift (red shift): Movement of λmax to longer wavelengths, often caused by electron-donating auxochromes extending conjugation through resonance [6]
- Hypsochromic shift (blue shift): Movement of λmax to shorter wavelengths, sometimes caused by electron-withdrawing groups or disruption of conjugation [6]
- Hyperchromic effect: Increase in absorption intensity, often resulting from auxochromes that enhance the probability of electronic transitions [42]
- Hypochromic effect: Decrease in absorption intensity, potentially caused by steric hindrance or aggregation [42]
Interpretation of UV-Vis Spectra for Functional Group Identification
Systematic Approach to Spectrum Analysis
Interpreting UV-Vis spectra to identify chromophores and functional groups requires a structured methodology [42]:
Analyze the Overall Spectrum Pattern: Examine the number, position, and shape of absorption bands. Each chromophore typically contributes one or more absorption bands based on its possible electronic transitions. For example, a nitro group typically exhibits two distinct bands: a weak n→π* transition (350-400 nm) and a stronger π→π* transition (200-250 nm) [42].
Identify Chromophores and Functional Groups: Correlate observed absorption bands with known chromophore characteristics. Key regions include [42]:
- Below 200 nm: σ→σ* transitions in saturated hydrocarbons
- 200-250 nm: π→π* transitions in isolated alkenes, π→π* transitions in conjugated dienes
- 250-290 nm: π→π* transitions in aromatic systems
- 270-300 nm: n→π* transitions in carbonyl compounds
Analyze Spectral Shifts and Intensity Changes: Evaluate how conjugation, auxochromes, and solvent effects modify the base chromophore absorption. Extended conjugation causes bathochromic shifts, moving absorption to longer wavelengths [42] [5].
Correlate Spectral Features with Possible Structures: Compare observed absorption characteristics with reference data to develop plausible structural candidates that align with the spectral features [42].
The following workflow diagram illustrates this systematic approach to interpreting UV-Vis spectra for functional group identification:
Quantitative Analysis Using Beer-Lambert Law
The Beer-Lambert Law forms the foundation for quantitative analysis in UV-Vis spectroscopy, relating the absorption of light to the properties of the material through which the light is traveling [6] [46]. The mathematical expression is:
A = εbc
Where:
- A is the measured absorbance (dimensionless)
- ε is the molar absorptivity or extinction coefficient (L·molâ»Â¹Â·cmâ»Â¹)
- b is the path length through the sample (cm)
- c is the concentration of the absorbing species (mol/L) [6] [47]
This relationship enables researchers to determine unknown concentrations of chromophores in solution by measuring absorbance at the compound's λmax and applying a calibration curve generated from standards with known concentrations [42]. The linear relationship typically holds for absorbance values between 0.1 and 1.0, though deviations may occur at higher concentrations due to instrumental limitations or molecular interactions [42].
Experimental Methodologies and Protocols
Sample Preparation and Measurement
Proper sample preparation is critical for obtaining reliable UV-Vis spectra. The following protocol outlines standard procedures for analyzing bioactive molecules:
Solvent Selection: Choose a solvent that does not absorb significantly in the spectral region of interest. Common solvents include water, hexane, methanol, and acetonitrile. Avoid solvents with carbonyl groups (e.g., acetone) or other chromophores that might interfere with sample absorption [42].
Sample Concentration Optimization: Prepare samples at appropriate concentrations to ensure absorbance readings fall within the optimal range (0.1-1.0 absorbance units). This typically requires concentrations in the micromolar to millimolar range, depending on the molar absorptivity of the chromophore [42].
Solution Clarification: Ensure complete dissolution and clarity of samples. Particulate matter can scatter light, particularly at shorter wavelengths, increasing apparent absorbance and distorting measurements. Filter samples if necessary using compatible filters [42].
Cuvette Selection and Handling: Use optically matched cuvettes with appropriate path lengths (typically 1 cm). Ensure cuvettes are clean and free from scratches, fingerprints, or residues that could scatter or absorb light. Mismatched cuvettes between sample and reference can introduce systematic errors [42].
Baseline Correction: Collect a baseline spectrum using pure solvent in both reference and sample cuvettes to account for solvent absorption and instrument characteristics. Subtract this baseline from sample measurements [42].
Spectrum Acquisition: Set appropriate instrument parameters including scanning speed, slit width, and wavelength range. For quantitative work, measure absorbance at the specific λmax of the compound [42].
Troubleshooting Common Experimental Issues
Several factors can compromise the quality and reliability of UV-Vis spectra [42]:
Solvent Effects: Solvent polarity can induce solvatochromic shifts. Polar solvents may cause bathochromic shifts for π→π* transitions and hypsochromic shifts for n→π* transitions. Hydrogen-bonding solvents can interact with chromophores, further altering spectral features [42] [2].
Stray Light: Imperfect monochromators may allow stray light to reach the detector, reducing apparent absorbance readings at high concentrations and lowering quantitative accuracy [42].
Chemical Degradation: Some bioactive compounds may degrade during preparation or measurement, generating new chromophores or modifying existing ones. This introduces misleading spectral features that do not represent the original compound [42].
Multiple Absorbing Species: The presence of multiple chromophores with overlapping spectra can complicate interpretation, requiring mathematical deconvolution or additional analytical techniques to isolate individual contributions [42].
Essential Research Reagents and Materials
Successful UV-Vis analysis of bioactive molecules requires specific reagents and materials. The following table details essential components of the researcher's toolkit:
| Category | Specific Items | Function and Application Notes |
|---|---|---|
| Solvents | HPLC-grade water, hexane, methanol, acetonitrile, chloroform | Dissolve samples without interfering absorptions; choose based on sample solubility and spectral requirements [42]. |
| Cuvettes | Quartz (UV-Vis), optical glass (Vis only), disposable plastic | Contain samples during measurement; quartz essential for UV range, glass sufficient for visible only [42]. |
| Reference Standards | Potassium dichromate, holmium oxide filter | Verify wavelength accuracy and photometric scale of spectrophotometer [42]. |
| Chromogenic Reagents | p-Nitroanilide (pNA), p-Nitrophenyl ester (ONp), Thiobenzyl ester (SBzl) | Derivatization agents for detecting specific functional groups or enzyme activities [47]. |
| Buffer Systems | Phosphate, Tris, carbonate buffers | Maintain constant pH for pH-sensitive chromophores or biological molecules [47]. |
| Calibration Standards | Compound-specific certified reference materials | Establish calibration curves for quantitative analysis [42]. |
UV-Vis spectroscopy provides an indispensable tool for identifying chromophores and functional groups in bioactive molecules through the detection of characteristic electronic transitions. The systematic interpretation of spectral features—including absorption maxima, band intensity, and shifts induced by conjugation or auxochromes—enables researchers to elucidate structural elements central to pharmaceutical development and biochemical analysis. When executed with proper experimental protocols and attention to potential artifacts, this technique yields valuable insights into molecular structure, concentration, and interactions that underpin drug discovery and development research.
Ultraviolet-visible (UV-Vis) spectroscopy serves as a fundamental analytical technique in pharmaceutical development, providing critical data on substance identity, strength, quality, and purity through the measurement of light absorption characteristics [48]. This technique operates on the principle that molecules absorb specific wavelengths of ultraviolet or visible light, corresponding to the energy required to promote electrons from their ground state to higher energy excited states [2]. The resulting absorption spectra provide characteristic fingerprints that can be leveraged for both qualitative identification and quantitative determination of drug compounds and their intermediates [48].
The theoretical foundation of UV-Vis spectroscopy centers on electronic transitions occurring when photons interact with molecular orbitals. When atoms or molecules absorb energy, electrons undergo promotion from their ground state to an excited state, with the specific energy requirements dictated by the molecular structure and bonding environment [2]. These electronic transitions typically involve the excitation of π, σ, and n electrons to anti-bonding orbitals (π* and σ), with the most analytically useful transitions (π→π and n→π*) occurring in the experimentally convenient wavelength range of 200-700 nm [2] [8]. The precise energy differences between molecular orbitals, and consequently the wavelengths of absorbed light, are influenced by the presence of chromophores—functional groups containing valence electrons of low excitation energy, such as C=C, C=O, and aromatic ring systems [2] [6].
Theoretical Foundations: Electronic Transitions and the Beer-Lambert Law
Fundamental Electronic Transitions in Organic Molecules
The absorption of UV or visible radiation corresponds to the excitation of outer electrons, with several distinct transition types providing analytical information [2]. Table 1 summarizes the primary electronic transitions relevant to pharmaceutical analysis.
Table 1: Characteristics of Electronic Transitions in UV-Vis Spectroscopy
| Transition Type | Energy Requirement | Typical Wavelength Range | Molar Absorptivity (ε, L·molâ»Â¹Â·cmâ»Â¹) | Chromophores Involved |
|---|---|---|---|---|
| σ → σ* | High | <200 nm | - | C-C, C-H single bonds |
| n → σ* | Moderate | 150-250 nm | Low | Saturated compounds with lone pairs |
| π → π* | Moderate to Low | 200-700 nm | 1,000-10,000 | Alkenes, alkynes, aromatics |
| n → π* | Low | 200-700 nm | 10-100 | Carbonyl, nitro compounds |
| Charge-transfer | Variable | 200-700 nm | >10,000 | Electron donor-acceptor complexes |
For drug molecules, the most analytically useful transitions involve π→π* and n→π* excitations due to their occurrence within the readily accessible UV-Vis range [2]. The presence of conjugation—alternating single and multiple bonds—extends electron delocalization, reducing the energy gap between molecular orbitals and resulting in absorption at longer wavelengths [6]. This phenomenon explains why many pharmaceutical compounds with conjugated systems exhibit characteristic absorption in the visible region, making them suitable for quantitative analysis [6].
The Beer-Lambert Law: Mathematical Foundation
The Beer-Lambert Law establishes the fundamental relationship between light absorption and solution properties, forming the cornerstone of quantitative UV-Vis analysis [38] [49]. This law states that the absorbance of light by a solution is directly proportional to the concentration of the absorbing species and the path length through which light travels [38] [39]. The mathematical expression of this relationship is:
A = ε · b · c
Where:
- A is absorbance (unitless)
- ε is the molar absorptivity coefficient (L·molâ»Â¹Â·cmâ»Â¹)
- b is the path length of light through the solution (cm)
- c is the concentration of the absorbing species (mol·Lâ»Â¹) [38] [39]
Absorbance relates to the intensity of incident (Iâ‚€) and transmitted (I) light according to the logarithmic relationship:
A = logâ‚â‚€(Iâ‚€/I) [38] [39]
Transmittance (T), defined as T = I/Iâ‚€, provides an alternative measurement of light attenuation, with absorbance and transmittance related through the equation:
A = -logâ‚â‚€(T) [39]
Table 2: Relationship Between Absorbance and Transmittance
| Absorbance (A) | Transmittance (T) | % Transmittance | Light Absorbed |
|---|---|---|---|
| 0 | 1 | 100% | 0% |
| 0.3 | 0.5 | 50% | 50% |
| 1 | 0.1 | 10% | 90% |
| 2 | 0.01 | 1% | 99% |
| 3 | 0.001 | 0.1% | 99.9% |
The molar absorptivity coefficient (ε) represents the intrinsic absorption strength of a compound at a specific wavelength, with higher values indicating greater absorption sensitivity [38]. This parameter is particularly valuable for method validation as it is theoretically independent of instrument and concentration conditions [38] [39].
Figure 1: Relationship between electronic transitions and quantitative analysis using the Beer-Lambert Law in pharmaceutical applications.
Practical Application in Drug Development
Quantitative Analysis of Active Pharmaceutical Ingredients
The application of Beer-Lambert Law enables precise quantification of drug concentrations throughout development and manufacturing processes. By measuring absorbance at characteristic wavelengths and applying the relationship A = εbc, researchers can determine unknown concentrations of active pharmaceutical ingredients (APIs) in solutions [39]. This approach requires establishing a calibration curve using standard solutions of known concentration, which demonstrates the linear relationship between absorbance and concentration as dictated by the Beer-Lambert Law [39].
A representative example of this application involves the quantification of Rhodamine B in aqueous solutions, where absorption spectra are measured across a range of known concentrations [39]. Plotting absorbance versus concentration at the wavelength of maximum absorption (λmax) yields a linear calibration curve, enabling the determination of unknown concentrations through interpolation [39]. This method provides the foundation for API quantification in various pharmaceutical formulations, with the linear relationship typically holding for solutions with concentrations below 0.01 M [6].
Case Study: Residual Solvent Analysis in Nanoparticulate Drug Delivery Systems
UV-Vis spectrophotometry plays a critical role in quality control during the development of advanced drug delivery systems [50]. Nanoparticle preparation methods frequently employ organic solvents such as acetone to solubilize non-polar constituents, potentially leaving residual solvent concentrations that may cause undesired effects during biological testing [50].
A validated UV-Vis spectroscopic method has been developed for direct quantification of residual acetone in nanoparticles suspensions, employing a derivatization reaction with vanillin to enhance detection sensitivity [50]. The optimized protocol demonstrates excellent analytical performance with the following validated parameters:
Table 3: Validation Parameters for UV-Vis Quantification of Residual Acetone in Nanoparticles
| Validation Parameter | Result | Acceptance Criteria |
|---|---|---|
| Linear Range | 10-50 μg/mL | R² ≥ 0.990 |
| Correlation Coefficient (R²) | 0.998 | R² ≥ 0.990 |
| Limit of Detection (LOD) | 2.6 μg/mL | - |
| Limit of Quantification (LOQ) | 7.8 μg/mL | - |
| Accuracy | Concluded "accurate" | Within specified range |
| Precision | Concluded "precise" | RSD ≤ 2% |
This method remains effective even in the presence of various surfactants commonly employed during nanoparticle preparation, demonstrating robustness for pharmaceutical quality control applications [50]. The approach exemplifies how UV-Vis spectroscopy combined with the Beer-Lambert Law provides a simple, inexpensive, yet reliable analytical technique for ensuring product safety in advanced drug delivery systems.
Experimental Protocols and Methodologies
Standard Operating Procedure for Drug Quantification
Protocol 1: Calibration Curve Method for API Quantification
Instrument Calibration:
Standard Solution Preparation:
- Prepare a stock solution of the reference standard drug compound in appropriate solvent.
- Create a series of standard solutions with known concentrations through serial dilution.
- Ensure concentrations fall within the linear range of the Beer-Lambert relationship (typically below 0.01 M) [6].
Spectral Acquisition:
Calibration Curve Construction:
- Plot absorbance versus concentration for standard solutions.
- Perform linear regression to obtain the equation: A = εbC + intercept.
- Verify linearity with correlation coefficient R² ≥ 0.995 [39].
Sample Analysis:
- Measure absorbance of unknown samples at the same λmax.
- Calculate concentration using the calibration curve equation.
Residual Solvent Quantification Protocol
Protocol 2: Derivative Spectrophotometry for Residual Acetone [50]
Sample Derivatization:
- Mix nanoparticle suspension with vanillin reagent under optimized conditions.
- Allow color development for specified time period.
Analysis Conditions:
- Use central composite design for method optimization.
- Measure absorbance at characteristic wavelength for acetone-vanillin complex.
- Apply correction factors determined during method validation.
Quantification:
- Calculate acetone concentration using pre-established calibration curve.
- Apply correction factors for surfactant interference when necessary.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 4: Essential Materials for UV-Vis Quantitative Analysis in Drug Development
| Item | Specification | Function |
|---|---|---|
| UV-Vis Spectrophotometer | Dual-beam preferred | Measures light absorption by samples [8] |
| Cuvettes | Quartz for UV range (200-400 nm) | Sample holder with defined path length [8] |
| Reference Standard | High-purity API compound | Establishing calibration curves [48] |
| Solvents | HPLC grade, UV-transparent | Dissolving samples without interfering absorption [8] |
| Volumetric Flasks | Class A, various volumes | Precise solution preparation [48] |
| Micropipettes | Calibrated, appropriate volume range | Accurate liquid handling for serial dilutions |
| Derivatization Reagents | e.g., Vanillin for acetone detection | Enhancing detection sensitivity for specific analytes [50] |
Method Validation and Regulatory Considerations
For pharmaceutical applications, UV-Vis methods must undergo comprehensive validation to ensure reliability, accuracy, and reproducibility [48] [50]. Key validation parameters include:
- Linearity: Demonstration of direct proportionality between absorbance and concentration across the specified range, typically with correlation coefficient R² ≥ 0.990 [50].
- Accuracy: Determination through recovery studies using spiked samples, with acceptable recovery generally between 98-102% [50].
- Precision: Evaluation through repeatability (intra-day) and intermediate precision (inter-day) studies, with relative standard deviation (RSD) typically ≤2% [50].
- Specificity: Ability to unequivocally assess the analyte in the presence of expected impurities, excipients, or derivatization agents [48].
- Limit of Detection (LOD) and Quantification (LOQ): Determination of the lowest detectable and quantifiable analyte concentrations, established based on signal-to-noise ratios [50].
The validated method for residual acetone quantification exemplifies this approach, having been optimized using central composite designs and validated according to international guidelines [50]. This rigorous validation ensures the method's suitability for its intended application in pharmaceutical quality control.
The integration of electronic transition theory with the Beer-Lambert Law provides a robust foundation for quantitative analysis throughout drug development processes. From API quantification in formulation studies to residual solvent analysis in advanced delivery systems, UV-Vis spectroscopy serves as an accessible, cost-effective, and reliable analytical technique. The direct proportionality between absorbance and concentration established by the Beer-Lambert Law enables precise quantification, while understanding electronic transitions facilitates method development and optimization. As pharmaceutical systems grow increasingly complex, the fundamental principles of UV-Vis spectroscopy remain essential tools for ensuring drug identity, strength, quality, and purity.
Monitoring Molecular Interactions and Reaction Kinetics with UV-Vis
Ultraviolet-visible (UV-Vis) spectroscopy is an indispensable analytical technique in modern research and drug development for monitoring molecular interactions and quantifying reaction kinetics. This methodology operates on a fundamental principle: it measures the amount of discrete wavelengths of UV or visible light that are absorbed by or transmitted through a sample in comparison to a reference or blank sample [8]. The technique is grounded in the basic theory of electronic transitions, which occur when molecules absorb light energy, prompting electrons to jump from a ground state to a higher energy, excited state [51]. The energy required for this transition is inversely proportional to the wavelength of light, meaning shorter wavelengths carry more energy [8]. The specific amount of energy needed is characteristic of a molecule's electronic structure, providing a fingerprint that can be used for identification and quantification [8].
The most common electronic transitions involve the promotion of electrons from the Highest Occupied Molecular Orbital (HOMO) to the Lowest Unoccupied Molecular Orbital (LUMO) [6] [1]. The energy difference between these orbitals determines the wavelength of light absorbed [6]. Molecules or parts of molecules that absorb light strongly in the UV-visible region are called chromophores [1]. Common chromophores include C=C, C=O, and aromatic rings, with their specific structure and degree of conjugation directly influencing the energy of the electronic transition and thus the observed absorption spectrum [6]. This foundational principle enables researchers to probe intricate molecular interactions, monitor the progression of chemical reactions, and determine kinetic parameters with precision.
Theoretical Foundations: From Electronic Transitions to Quantitative Analysis
Chromophores, Auxochromes, and Spectral Interpretation
The absorption of UV-Vis light by a molecule is primarily dictated by its chromophores. Conjugation, where alternating single and double bonds extends electron delocalization across multiple atoms, has a profound effect on electronic transitions. Increased conjugation leads to a smaller energy gap between the HOMO and LUMO, resulting in absorption at longer wavelengths—a phenomenon known as a bathochromic shift (or red shift) [6]. For example, beta-carotene, with its system of 11 conjugated double bonds, absorbs light in the blue region of the visible spectrum, which is why carrots appear orange [1].
The absorption properties of chromophores can be further modified by auxochromes. These functional groups, which typically contain lone pairs of electrons (e.g., -OH, -NHâ‚‚), can attach to a chromophore and alter the wavelength and intensity of absorption. Electron-donating auxochromes often cause a bathochromic shift, while electron-withdrawing groups can lead to a hypsochromic shift (blue shift), which is a shift to shorter wavelengths [6]. The ability to predict and interpret these shifts is crucial for understanding how molecular structure changes during a reaction or interaction.
The Beer-Lambert Law and Quantitation
The cornerstone of quantitative analysis in UV-Vis spectroscopy is the Beer-Lambert Law. This law establishes a linear relationship between absorbance, the concentration of the absorbing species, and the path length the light travels through the sample [51] [6]. It is mathematically expressed as: [ A = \varepsilon b c ] Where:
- A is the measured absorbance (unitless)
- ε is the molar absorptivity (or extinction coefficient) with typical units of L molâ»Â¹ cmâ»Â¹
- b is the path length of the cuvette (cm)
- c is the concentration of the analyte (mol Lâ»Â¹) [51]
The molar absorptivity (ε) is a critical parameter that measures how strongly a substance absorbs light at a specific wavelength. It relates directly to the probability of the electronic transition; allowed transitions have high molar absorptivities (>10,000 L molâ»Â¹ cmâ»Â¹), while forbidden transitions have low values (<100 L molâ»Â¹ cmâ»Â¹) [6]. For accurate quantitation, absorbance values should ideally be kept below 1 to remain within the instrument's dynamic range, as an absorbance of 1 means 90% of the incoming light has been absorbed, leaving little light for the detector to measure reliably [8]. This can be achieved by diluting the sample or using a cuvette with a shorter path length.
Table 1: Key Spectral Shifts and Their Molecular Origins
| Shift Type | Description | Common Causes |
|---|---|---|
| Bathochromic (Red) | Shift to longer wavelength (lower energy) | Increased conjugation, electron-donating groups, solvent polarity, pH changes [6] |
| Hypsochromic (Blue) | Shift to shorter wavelength (higher energy) | Decreased conjugation, electron-withdrawing groups, solvent effects, pH changes [6] |
| Hyperchromic | Increase in absorption intensity | Structural changes that increase the probability of the transition |
| Hypochromic | Decrease in absorption intensity | Structural changes that decrease the probability of the transition [6] |
Instrumentation and Experimental Design
Core Components of a UV-Vis Spectrophotometer
A UV-Vis spectrophotometer, regardless of its configuration, is built around several key components that work in concert to acquire absorption data [8] [51].
- Light Source: A stable source emitting light across a wide range of wavelengths is essential. Instruments often use a combination of lamps: a deuterium lamp for the UV region and a tungsten or halogen lamp for the visible region. The instrument switches between these sources during a scan, typically between 300 and 350 nm [8].
- Wavelength Selector: This component selects specific wavelengths from the broad spectrum emitted by the source. The most versatile selector is a monochromator, which uses a diffraction grating to separate light into a narrow band of wavelengths. Filters (absorption, interference, cutoff, bandpass) may also be used, often in conjunction with a monochromator, to further refine the wavelength selection and improve the signal-to-noise ratio [8].
- Sample Holder: Samples are typically analyzed in solution held within a cuvette. For UV studies, quartz cuvettes are mandatory as they are transparent to most UV light. Glass and plastic cuvettes are inappropriate for UV because they absorb UV light [8]. Cuvette-free systems are also available for analyzing very small sample volumes, such as in DNA or RNA analysis [8].
- Detector: After the light passes through the sample, a detector converts it into an electronic signal. Common detectors include photomultiplier tubes (PMTs), which are excellent for detecting very low light levels, and semiconductor-based detectors like photodiodes and charge-coupled devices (CCDs) [8].
The configuration of these components can vary. Single-beam instruments use one light path, passing through a single sample at a time. Double-beam instruments split the light, allowing simultaneous measurement of the sample and a reference, which provides more accurate readings by compensating for source fluctuations [51]. Simultaneous or diode-array instruments have no moving monochromator; instead, they disperse the light after it passes through the sample onto a CCD array, allowing the detection of all wavelengths at once, which greatly increases speed [51].
Essential Research Reagent Solutions and Materials
The reliability of UV-Vis data is highly dependent on the quality and appropriateness of the materials and reagents used. The following table details key components for a successful experiment.
Table 2: Essential Research Reagents and Materials for UV-Vis Spectroscopy
| Item | Function/Description | Key Considerations |
|---|---|---|
| Quartz Cuvettes | Holds liquid sample for analysis. | Required for UV range (transparent down to ~200 nm). Standard path length is 1 cm [8]. |
| Solvents (e.g., Water, Hexanes, Methanol) | Dissolves the analyte to form a homogeneous solution. | Must be high-purity (UV-grade) and transparent in the spectral region of interest. The solvent for the sample and reference must be identical [51]. |
| Buffers (e.g., Phosphate, Tris) | Maintains constant pH, crucial for stabilizing biomolecules. | Must not absorb significantly at the wavelengths being monitored. The buffer without the analyte is used for the blank/reference [8]. |
| Standard Solutions | Solutions of known concentration for creating a calibration curve. | Used for quantitative work. Must be prepared accurately with volumetric glassware; typically, 3-5 concentrations are needed for a reliable curve [51]. |
| D-luciferin (for bioluminescence BRET) | Substrate for luciferase enzyme in advanced bioluminescence probes. | Used in specialized applications like NIR-II bioluminescence for in vivo imaging, enabling reaction monitoring without external light [52]. |
Monitoring Reaction Kinetics: Protocols and Data Analysis
Experimental Protocol for Kinetic Monitoring
UV-Vis spectroscopy is a powerful tool for monitoring chemical reactions in real-time, providing insights into reaction mechanisms and kinetics [53]. The following is a detailed methodology for a typical reaction kinetics experiment.
- Identify the Analytic Wavelength: Prior to the kinetic experiment, obtain a full absorbance spectrum of the reactant, product, or a key intermediate. The wavelength of maximum absorbance (λ_max) for the species being tracked is typically chosen for monitoring, as this provides the greatest sensitivity [1].
- Prepare Calibration Standards (for quantitative analysis): If the goal is to determine concentration changes quantitatively, prepare a series of standard solutions with known concentrations of the target analyte. Plot absorbance versus concentration to create a calibration curve that obeys the Beer-Lambert law. A correlation coefficient (R²) of 0.9 or better is typically considered acceptable [51].
- Initialize the Reaction: In a suitable reaction vessel (which may be a cuvette itself), mix the reactants to initiate the reaction. For rapid reactions, stopped-flow accessories may be required. Ensure the solvent and conditions (temperature, pH) are consistent and controlled.
- Acquire Time-Dependent Data: Configure the spectrophotometer to measure absorbance at the pre-determined analytic wavelength at regular time intervals. The instrument will track the change in absorbance as the reaction progresses. For reactions where multiple species evolve, full spectra can be collected at each time point [53].
- Data Processing: Convert the recorded absorbance values into concentration values using the calibration curve or the known molar absorptivity (ε) of the species. Plot concentration versus time to generate the kinetic profile of the reaction.
Determining Reaction Order and Rate Constants
The analysis of the concentration-time data allows for the determination of the reaction order and the calculation of the rate constant. The following diagram and table outline this analytical workflow.
Table 3: Kinetic Analysis Methods for Common Reaction Orders
| Reaction Order | Integrated Rate Law | Linear Plot | Slope & Y-Intercept | Half-Life Expression |
|---|---|---|---|---|
| Zero Order | ( [A]t = -kt + [A]0 ) | [A] vs. Time | Slope = -k, Y-int = [A]â‚€ | ( t{1/2} = \frac{[A]0}{2k} ) |
| First Order | ( \ln[A]t = -kt + \ln[A]0 ) | ln[A] vs. Time | Slope = -k, Y-int = ln[A]â‚€ | ( t_{1/2} = \frac{\ln(2)}{k} ) |
| Second Order | ( \frac{1}{[A]t} = kt + \frac{1}{[A]0} ) | 1/[A] vs. Time | Slope = +k, Y-int = 1/[A]â‚€ | ( t{1/2} = \frac{1}{k[A]0} ) |
The reaction order is identified by testing which plot (concentration, natural log of concentration, or inverse concentration versus time) yields the straightest line (highest R² value). The rate constant k is then derived directly from the slope of the corresponding linear plot [53]. Environmental factors such as temperature, pH, and ionic strength can significantly impact the observed reaction rate, and their effects can be systematically studied using this protocol [53].
Advanced Applications: Molecular Interactions and In Vivo Imaging
The application of UV-Vis spectroscopy extends far beyond simple kinetic studies of homogeneous solutions. It is a versatile technique for probing a variety of molecular interactions.
Biomolecular Binding Studies: UV-Vis can be used to study the binding of small molecules to DNA or proteins. Intercalation of a drug molecule into DNA, for instance, often causes a bathochromic shift and hypochromicity in the DNA's absorption spectrum, which can be used to calculate binding constants.
Monitoring Enzyme-Substrate Complexes: The formation and decay of enzyme-substrate intermediates can often be tracked if their absorption spectra differ from the starting materials. This provides direct insight into enzymatic mechanisms.
Emerging Frontiers: NIR-II Bioluminescence Imaging: A cutting-edge advancement is the development of bioluminescence probes (BPs) that operate in the second near-infrared window (NIR-II, 1000-1700 nm). These probes use a Bioluminescence Resonance Energy Transfer (BRET) process, coupled with a multi-step Fluorescence Resonance Energy Transfer (FRET) cascade, to shift the emission of a luciferase enzyme from the visible range to 1029 nm [52]. This allows for high-contrast in vivo imaging with significantly higher signal-to-noise ratios and spatial resolution compared to conventional visible light bioluminescence or NIR-I fluorescence imaging, as scattering and tissue absorption are minimized in the NIR-II region [52]. This technology has been successfully applied to image vasculature, lymphatics, and even ATP-mediated tumor metastasis in live mice, opening new avenues for monitoring biological reactions and interactions directly within living organisms [52].
UV-Vis spectroscopy, firmly rooted in the principles of electronic transitions, remains a cornerstone technique for monitoring molecular interactions and reaction kinetics. Its ability to provide both qualitative and quantitative data in real-time makes it invaluable for researchers and drug development professionals. From foundational studies in solution to the emerging frontier of in vivo NIR-II bioluminescence imaging, the continued evolution of UV-Vis methodologies ensures its central role in advancing scientific discovery and therapeutic innovation. By adhering to rigorous experimental protocols, such as proper calibration and material selection, and by applying robust kinetic analysis frameworks, scientists can extract profound insights into the dynamics and mechanisms of molecular processes.
This whitepaper presents a comprehensive technical examination of bakuchiol quantification within cosmetic and pharmaceutical matrices, framed through the theoretical lens of electronic transitions in Ultraviolet-Visible (UV-Vis) spectroscopy. As a natural retinoid alternative, bakuchiol presents specific analytical challenges that we explore through comparative method evaluation. The study details experimental protocols for UV-Vis spectroscopy, High-Performance Liquid Chromatography (HPLC), and Nuclear Magnetic Resonance (NMR) spectroscopy, providing quantitative comparisons of accuracy, sensitivity, and operational efficiency. Within the context of electronic transition theory, we elucidate the molecular basis for bakuchiol's characteristic absorption profile at 262 nm, establishing how π→π* transitions within its conjugated system facilitate detection and quantification. The findings demonstrate that UV-Vis spectroscopy offers a rapid screening method, while HPLC and 1H qNMR provide superior quantification, with NMR achieving comparable accuracy to HPLC with significantly reduced analysis time. This research provides formulation scientists and analytical chemists with validated methodologies for quality control, emphasizing the fundamental spectroscopic principles that underpin these analytical techniques.
Bakuchiol as a Bioactive Compound
Bakuchiol (C22H34O2) is a meroterpene natural product first isolated from the seeds of Psoralea corylifolia in 1966 [54]. It has gained significant prominence in cosmetic and pharmaceutical formulations as a natural alternative to retinoids, offering comparable anti-aging benefits through antioxidant activity and stimulation of retinoic acid receptor (RAR) gene expression without the associated adverse effects like skin irritation and phototoxicity [54]. The naturally occurring stereoisomer is S-(+)-bakuchiol, which is optically active and typically constitutes 1-7% of the dry weight of Psoralea corylifolia fruits [54]. Its mechanism of action is not yet fully elucidated, necessitating robust analytical methods for quality control in commercial products where efficacy claims must be substantiated.
Analytical Chemistry Context
The quantification of active ingredients in complex matrices like cosmetic formulations presents significant challenges due to interference from excipients, emulsifiers, preservatives, and other formulation components. Effective quality control requires methods that are specific, accurate, precise, and efficient for routine analysis. This case study examines three principal techniques—UV-Vis spectroscopy, HPLC, and NMR spectroscopy—contextualizing their application for bakuchiol quantification within the fundamental theory of molecular electronic transitions observed in UV-Vis spectroscopy [55]. This theoretical framework provides the basis for understanding light absorption behavior and its application to analytical quantification.
Theoretical Framework: Electronic Transitions in UV-Vis Spectroscopy
UV-Vis spectroscopy probes the electronic structure of molecules by measuring their absorption of electromagnetic radiation in the ultraviolet (200-400 nm) and visible (400-800 nm) regions [56]. When a molecule absorbs light energy corresponding to the energy difference between its ground and excited electronic states, valence electrons are promoted from their ground state orbitals to higher energy excited state orbitals [4]. The energy of absorbed photons is quantized, following the equation E = hc/λ, where E is energy, h is Planck's constant, c is the speed of light, and λ is wavelength [57] [4]. This relationship forms the theoretical basis for all UV-Vis spectroscopic analysis.
The absorption process promotes electrons from the Highest Occupied Molecular Orbital (HOMO) to the Lowest Unoccupied Molecular Orbital (LUMO), with the energy difference (band gap) typically ranging from 125 to 650 kJ/mol for transitions observed in the UV-Vis region [58]. In molecular orbital theory, the energy levels follow this general order: σ orbitals (lowest energy) < π orbitals < n (non-bonding) orbitals < π* orbitals < σ* orbitals (highest energy) [59] [58].
Relevant Electronic Transitions for Organic Analysis
For organic molecules like bakuchiol, several types of electronic transitions are theoretically possible, but only specific transitions are relevant for practical UV-Vis spectroscopy in the 200-800 nm range:
π→π* Transitions: These transitions occur in molecules with unsaturated centers (e.g., alkenes, aromatics, carbonyl compounds) where electrons in Ï€ bonding orbitals are excited to Ï€* antibonding orbitals [59]. They typically require less energy than σ→σ* transitions, with absorption bands appearing between 170-190 nm for unconjugated alkenes [59]. π→π* transitions are "allowed transitions" with high probability, resulting in high molar absorptivity (εmax > 1000 L·molâ»Â¹Â·cmâ»Â¹) [2]. In conjugated systems, these transitions shift to longer wavelengths (bathochromic shift) with increased intensity.
n→π* Transitions: These transitions occur when non-bonding electrons (lone pairs) on heteroatoms (like oxygen in carbonyl groups) are excited to Ï€* antibonding orbitals [59] [2]. They involve the least energy among common transitions, resulting in absorption at longer wavelengths (e.g., ~280 nm for saturated aliphatic ketones) [59]. However, n→π* transitions are "forbidden transitions" by symmetry considerations, resulting in low intensity bands with molar absorptivities typically between 10-100 L·molâ»Â¹Â·cmâ»Â¹ [59] [2].
n→σ* Transitions: These transitions occur in saturated compounds containing heteroatoms with lone pairs (e.g., oxygen, nitrogen, sulfur) and typically absorb in the 150-250 nm range [2]. They generally have lower energy requirements than σ→σ* transitions but are less relevant for conjugated systems like bakuchiol.
For analytical chemistry applications, π→π* and n→π* transitions are most practically useful, as their energy differences correspond to wavelengths within the conveniently measurable UV-Vis range (200-400 nm) [59]. The extent of conjugation in a molecule directly affects the energy gap for π→π* transitions; increased conjugation decreases the HOMO-LUMO gap, resulting in absorption at longer wavelengths (red shift) [57] [4].
Table 1: Characteristics of Electronic Transitions in UV-Vis Spectroscopy
| Transition Type | Energy Requirement | Typical Wavelength Range | Molar Absorptivity (ε) | Chromophores Involved |
|---|---|---|---|---|
| σ→σ* | Highest | <200 nm | High | C-C, C-H single bonds |
| n→σ* | High | 150-250 nm | Low | O, N, S with lone pairs |
| π→π* | Medium | 170-250 nm (unconjugated); longer when conjugated | High (1,000-10,000) | Alkenes, alkynes, aromatics, carbonyls |
| n→π* | Lowest | 250-400 nm | Low (10-100) | Carbonyls, nitro compounds |
Solvent Effects on Electronic Transitions
The solvent in which analysis is conducted significantly affects absorption spectra. For n→π* transitions, increasing solvent polarity causes a shift to shorter wavelengths (blue shift or hypsochromic shift) due to increased solvation of lone pairs, which stabilizes the n orbital more than the π* orbital [2]. Conversely, π→π* transitions often experience a small shift to longer wavelengths (red shift or bathochromic shift) with increasing solvent polarity due to greater stabilization of the excited state relative to the ground state [2]. These solvent effects must be considered when developing analytical methods.
Bakuchiol-Specific Electronic Transitions and Spectral Characteristics
Molecular Structure and Chromophores
Bakuchiol contains a conjugated phenolic structure with an isoprenoid side chain, featuring several chromophores that contribute to its UV absorption characteristics [54]. The para-substituted phenolic ring with extended conjugation through the isoprenoid chain creates a significant π-electron system that dominates its UV absorption profile [54]. This conjugated system allows for π→π* transitions with energy differences corresponding to wavelengths in the easily measurable UV range.
Characteristic Absorption Profile
Bakuchiol exhibits a characteristic UV absorption maximum at λmax = 262 nm in ethanol [54]. This absorption band arises primarily from π→π* electronic transitions within the conjugated system of the molecule [54]. The extensive conjugation across the phenolic ring and isoprenoid side chain lowers the energy gap between the HOMO and LUMO orbitals, resulting in absorption at this relatively long wavelength in the UV region [57] [4]. The absorption at 262 nm is consistent with allowed transitions, exhibiting sufficiently high molar absorptivity for reliable quantification in analytical applications.
The following diagram illustrates the electronic transition in bakuchiol responsible for its characteristic UV absorption, showing the promotion of an electron from the HOMO to the LUMO upon absorption of a 262 nm photon:
Diagram 1: Electronic transition of bakuchiol at 262 nm
Experimental Protocols for Bakuchiol Quantification
UV-Vis Spectrophotometric Method
Principle
This method relies on the direct proportionality between absorbance and concentration described by Beer-Lambert Law (A = εbc), where A is absorbance, ε is molar absorptivity, b is path length, and c is concentration [2]. Bakuchiol's strong absorption at 262 nm due to π→π* electronic transitions enables its quantification in solution [54].
Materials and Reagents
- Pure bakuchiol standard (>98% purity)
- Ethanol (absolute, spectroscopic grade)
- Cosmetic formulations containing bakuchiol
- Volumetric flasks (10 mL, 25 mL)
- Micropipettes (100-1000 μL)
- UV-Vis spectrophotometer with quartz cuvettes (1 cm path length)
Procedure
Standard Solution Preparation: Accurately weigh approximately 10 mg of pure bakuchiol standard and dissolve in ethanol in a 25 mL volumetric flask. Dilute to mark with ethanol to create a stock solution of approximately 400 μg/mL.
Calibration Curve Standards: Prepare a series of standard solutions by diluting the stock solution with ethanol to concentrations of 5, 10, 20, 40, and 80 μg/mL.
Sample Preparation: For oil-based formulations, accurately weigh approximately 50 mg of product and dissolve in 25 mL ethanol. For emulsion-based formulations, attempt complete dissolution in ethanol, though this may be challenging with oil-in-water emulsions [54]. Sonicate for 10 minutes and centrifuge if necessary to obtain a clear supernatant.
Spectral Acquisition: Measure absorbance of all standards and samples from 200-400 nm against an ethanol blank. Record absorbance at λmax = 262 nm.
Quantification: Construct a calibration curve by plotting absorbance at 262 nm versus standard concentrations. Determine sample concentration from the linear regression equation of the calibration curve.
Method Validation
- Linearity: Establish over concentration range of 5-80 μg/mL with correlation coefficient (R²) >0.995
- Limit of Detection (LOD): Approximately 1-2 μg/mL
- Limit of Quantification (LOQ): Approximately 5 μg/mL
- Precision: %RSD <2.5% for repeatability
High-Performance Liquid Chromatography (HPLC) Method
Principle
HPLC separates bakuchiol from other formulation components using a reverse-phase C18 column with isocratic elution, followed by detection at 260 nm based on bakuchiol's π→π* electronic transition characteristics [54] [60].
Materials and Reagents
- HPLC system with DAD detector
- Reverse-phase C18 column (250 × 4.6 mm, 5 μm)
- Acetonitrile (HPLC grade)
- Formic acid (analytical grade)
- Bakuchiol standard (>98% purity)
- Cosmetic samples
Procedure
- Mobile Phase: Acetonitrile with 1% formic acid
- Flow Rate: 1.0 mL/min
- Detection: 260 nm
- Injection Volume: 10-20 μL
- Column Temperature: 25°C
- Standard Preparation: Prepare bakuchiol standards in acetonitrile at concentrations of 5, 10, 25, 50, and 100 μg/mL
- Sample Preparation: Extract approximately 50 mg of cosmetic product in 25 mL acetonitrile, sonicate for 15 minutes, and filter through 0.45 μm membrane
- Quantification: Identify bakuchiol peak at RT ~31.8 minutes and quantify using external standard method [54]
Method Validation
- LOD: 0.5 μg/mL
- LOQ: 1.5 μg/mL
- Linearity: R² >0.999 over 5-100 μg/mL range
- Precision: %RSD <2.5% for intra-day variation
Quantitative ¹H NMR (qNMR) Method
Principle
This method uses nicotinamide as an internal standard to quantify bakuchiol by comparing the integral ratios of selected proton signals, leveraging the principle that signal intensity is directly proportional to the number of nuclei generating the signal [54].
Materials and Reagents
- NMR spectrometer (400 MHz or higher)
- Deuterated chloroform (CDCl₃)
- Nicotinamide (analytical standard)
- Bakuchiol standard
- Cosmetic samples
Procedure
- Internal Standard Solution: Prepare 5 mg/mL nicotinamide in CDCl₃
- Standard Preparation: Accurately weigh bakuchiol standard and dissolve in 600 μL of internal standard solution in an NMR tube
- Sample Preparation: Extract bakuchiol from approximately 50 mg cosmetic product with 600 μL internal standard solution in CDCl₃
- Spectral Acquisition: Acquire ¹H NMR spectra with sufficient relaxation delay (d1 > 5×T1)
- Quantification: Compare integrals of bakuchiol aromatic protons (δ = 7.25-7.20 ppm) or olefinic protons (δ = 6.10-6.00 ppm) with nicotinamide reference signals
Method Validation
- Specificity: Excellent structural confirmation through chemical shift assignment
- Accuracy: Comparable to HPLC with proper calibration
- Analysis Time: Significantly shorter than HPLC (approximately 5-10 minutes per sample after preparation) [54]
The following workflow diagram summarizes the key steps in bakuchiol analysis across the three analytical techniques:
Diagram 2: Workflow for bakuchiol quantification methods
Comparative Data Analysis
Quantitative Method Comparison
The following table summarizes the performance characteristics of the three analytical methods for bakuchiol quantification:
Table 2: Comparison of Analytical Methods for Bakuchiol Quantification
| Parameter | UV-Vis Spectroscopy | HPLC with UV Detection | ¹H qNMR |
|---|---|---|---|
| Principle | Electronic transitions (π→π*) at 262 nm | Separation + electronic transition detection at 260 nm | Proton signal integration relative to internal standard |
| Analysis Time | ~30 minutes | ~45 minutes/sample | ~10 minutes/sample (after preparation) |
| LOD | 1-2 μg/mL | 0.5 μg/mL | ~5 μg/mL |
| LOQ | 5 μg/mL | 1.5 μg/mL | ~15 μg/mL |
| Precision (%RSD) | <3% | <2.5% | <2% |
| Linearity (R²) | >0.995 | >0.999 | >0.999 |
| Specificity | Low (interference from other UV absorbers) | High (separation prior to detection) | Very High (structural confirmation) |
| Sample Preparation | Simple dissolution in ethanol | Extraction and filtration | Dissolution in deuterated solvent |
| Key Advantage | Rapid, inexpensive | Specific, sensitive, widely available | Structural confirmation, minimal calibration |
| Key Limitation | Limited specificity for complex matrices | Longer analysis time, solvent consumption | Lower sensitivity, specialized equipment |
Application to Commercial Product Analysis
In a study analyzing six commercial cosmetic serums, these methods demonstrated varying effectiveness [54]:
UV-Vis Analysis: Successfully quantified bakuchiol in oil-based formulations (samples 1, 3, and 4) but encountered challenges with oil-in-water emulsions (samples 5 and 6) where complete dissolution was problematic [54]. Sample 2 showed no bakuchiol detection despite manufacturer claims.
HPLC Analysis: Confirmed UV-Vis findings with higher specificity, detecting no bakuchiol in sample 2 and quantifying sample 4 at 3.6% bakuchiol content—the highest among tested products [54]. Sample 3 matched its labeled concentration at 1%, while sample 1 contained only 0.51% (approximately 50% of declared content) [54].
qNMR Analysis: Provided comparable results to HPLC with significantly shorter analysis time, making it suitable for routine quality control [54]. Successfully detected bakuchiol in samples where it was present, with structural confirmation through characteristic aromatic (δ = 7.25-7.20 ppm) and olefinic (δ = 6.10-6.00 ppm) proton signals [54].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential Research Reagents and Materials for Bakuchiol Analysis
| Item | Specification | Function in Analysis |
|---|---|---|
| Bakuchiol Standard | >98% purity, S-(+)-enantiomer preferred | Primary reference standard for calibration and method validation |
| Ethanol (Absolute) | Spectroscopic grade, low UV absorbance | Solvent for UV-Vis analysis and standard preparation |
| Acetonitrile | HPLC grade, low UV cutoff | Mobile phase component for HPLC analysis |
| Deuterated Chloroform (CDCl₃) | 99.8% deuterium, TMS as internal standard | Solvent for NMR analysis providing deuterium lock signal |
| Nicotinamide | Analytical standard grade | Internal standard for qNMR quantification |
| Formic Acid | Analytical grade, >98% purity | Mobile phase modifier for HPLC to improve peak shape |
| C18 Reverse-Phase Column | 250 × 4.6 mm, 5 μm particle size, end-capped | Stationary phase for HPLC separation of bakuchiol from matrix components |
| Quartz Cuvettes | 1 cm path length, UV-transparent | Sample holder for UV-Vis spectroscopy measurements |
| Syringe Filters | 0.45 μm PTFE or nylon membrane | Filtration of HPLC samples to remove particulate matter |
| Deuterated NMR Solvents | CDCl₃, DMSO-d6, Methanol-d4 | Solvents for NMR analysis providing field frequency lock |
This technical examination demonstrates that effective quantification of bakuchiol in cosmetic and pharmaceutical formulations requires understanding both the analytical techniques and the fundamental electronic transition theory that underpins UV-based detection methods. Bakuchiol's characteristic absorption at 262 nm, resulting from π→π* electronic transitions within its conjugated system, provides the theoretical basis for its detection and quantification by UV-Vis spectroscopy and HPLC-UV.
The comparative analysis reveals that while UV-Vis spectroscopy offers a rapid, cost-effective screening method, its limitations in specificity make it less reliable for complex emulsion formulations. HPLC provides superior specificity and sensitivity, enabling accurate quantification even in complex matrices, though with longer analysis times. Remarkably, ¹H qNMR emerges as a compelling alternative, providing comparable accuracy to HPLC with significantly reduced analysis time and the added benefit of structural confirmation.
For researchers and quality control professionals, selection of the appropriate analytical method should be guided by specific application requirements: UV-Vis for rapid screening, HPLC for regulatory-grade quantification, and qNMR for high-throughput quality control with structural verification. All these techniques, however, ultimately rely on the fundamental principles of electronic transitions that govern how organic molecules like bakuchiol interact with ultraviolet radiation—demonstrating the enduring importance of spectroscopic theory in applied analytical chemistry.
Solving Spectral Puzzles: Troubleshooting Common UV-Vis Challenges
Avoiding Solvent Selection Errors and Managing Solvatochromic Shifts
In UV-Vis spectroscopy, electronic transitions occur when molecules absorb light, promoting electrons from ground-state to higher-energy excited-state orbitals [2]. The energy required for these transitions is not an intrinsic property of the molecule alone; it is profoundly influenced by the solvent environment. This phenomenon, known as solvatochromism, describes the shift in the absorption maximum (λmax) due to differential solvation of the ground and excited states [61] [62].
For researchers in drug development and chemical research, ignoring these effects can lead to inaccurate data interpretation, flawed compound identification, and poor reproducibility. This guide integrates the theory of electronic transitions with practical strategies for solvent selection and the management of solvatochromic shifts, ensuring robust and reliable spectroscopic analysis.
Core Theory: Electronic Transitions and Solvent Interaction
A foundational understanding of molecular orbitals and how solvents interact with them is crucial for predicting and managing solvatochromism.
Key Electronic Transitions in UV-Vis
The most common transitions in organic molecules involve the excitation of n (non-bonding), π (pi-bonding), and σ (sigma-bonding) electrons [2]. Their energy requirements and characteristics are summarized in Table 1.
Table 1: Common Electronic Transitions in UV-Vis Spectroscopy
| Transition Type | Typical Energy/Wavelength | Molar Absorptivity (ε) | Chromophore Example |
|---|---|---|---|
| σ → σ* | Very High / < 200 nm | High | C-C, C-H (Alkanes) |
| n → σ* | High / 150 - 250 nm | Low (10-100 L molâ»Â¹ cmâ»Â¹) | CH₃OH, CH₃NHâ‚‚ |
| Ï€ → Ï€* | Medium / 200 - 700 nm | High (1,000-10,000 L molâ»Â¹ cmâ»Â¹) | C=C, C=O (Conjugated) |
| n → Ï€* | Low / 270 - 300 nm | Low (10-100 L molâ»Â¹ cmâ»Â¹) | C=O, NOâ‚‚ |
The energy gap between the Highest Occupied Molecular Orbital (HOMO) and the Lowest Unoccupied Molecular Orbital (LUMO) determines the wavelength of absorbed light. Conjugation delocalizes π electrons, shrinking this HOMO-LUMO gap and causing a bathochromic (red) shift to longer wavelengths [6].
The Physical Basis of Solvatochromism
A solute's ground and excited states often have different electron distributions and dipole moments. A polar solvent will stabilize the state with the larger dipole moment more effectively. The direction of the spectral shift depends on which state is more stabilized [2] [62]:
- Negative Solvatochromism (Blue Shift/Hypsochromic Shift): Occurs when the ground state is more polar than the excited state. Increasing solvent polarity stabilizes the ground state more, increasing the energy gap (ΔE), thus shifting absorption to shorter wavelengths. This is typical for n→π* transitions, where the n-electron is stabilized by hydrogen-bonding in polar protic solvents [2].
- Positive Solvatochromism (Red Shift/Bathochromic Shift): Occurs when the excited state is more polar than the ground state. Increasing solvent polarity stabilizes the excited state more, decreasing the energy gap (ΔE), thus shifting absorption to longer wavelengths. This is common for π→π* and charge-transfer transitions [2] [6].
The following diagram illustrates these concepts and their relationship to the HOMO-LUMO energy gap.
A Practical Guide to Solvent Selection and Error Avoidance
Improper solvent choice is a primary source of error in UV-Vis spectroscopy, affecting both the accuracy and the very feasibility of a measurement.
Common Solvent Selection Errors and Consequences
- Absorbance in the Analyte's Wavelength Range: Using a solvent that itself absorbs significantly at the λmax of the analyte is a critical error. The solvent will compete for light absorption, reducing the dynamic range, distorting the absorption band, and leading to inaccurate concentration calculations via the Beer-Lambert Law [42] [8].
- Ignoring Solvent Polarity Effects: Failing to account for solvatochromism can lead to misidentification of compounds. A reported λmax that does not specify the solvent is scientifically useless, as the value can shift by tens of nanometers in different environments [42] [61].
- Overlooking Hydrogen-Bonding Potential: For chromophores with lone pairs (e.g., carbonyls, azo groups), protic solvents (water, alcohols) can cause significant blue shifts in n→π* transitions by hydrogen-bonding with the non-bonding electrons. This specific interaction can overshadow general polarity effects [2] [61].
- Chemical Reactivity and Sample Degradation: Some solvents can react with the analyte. For example, chlorinated solvents can participate in reactions with nucleophiles, and alkaline conditions can degrade sensitive molecules like reducing sugars [61].
Solvent Selection Checklist
To avoid these pitfalls, use the following checklist:
- Transparency Window: Confirm the solvent's UV-cutoff wavelength is at least 20-50 nm below your analyte's expected λmax [8].
- Polarity Matching: Consider the polarity of your analyte and the solvent. Document the solvent used for all measurements.
- Chemical Inertness: Ensure the solvent does not react with your analyte.
- Purity: Use solvents with high spectroscopic purity.
- Cuvette Compatibility: Remember that glass and plastic cuvettes absorb UV light. Use quartz cuvettes for work below ~350 nm [8].
Table 2: Properties of Common UV-Vis Solvents
| Solvent | UV Cutoff (nm)* | Polarity | Hydrogen-Bonding | Key Considerations |
|---|---|---|---|---|
| Water | ~190 | High | Protic | Excellent for polar biomolecules; high cutoff. |
| Acetonitrile | ~190 | High | Aprotic | Good for a wide range of polar compounds. |
| n-Hexane | ~200 | Low | Non | Good for non-polar analytes. |
| Methanol | ~205 | High | Protic | Can cause H-bonding shifts in n→π* transitions. |
| Chloroform | ~245 | Low | Non | Can contain stabilizing ethanol; reacts with some bases. |
| Acetone | ~330 | Medium | Aprotic | High cutoff makes it unsuitable for many UV analyses. |
| N,N-Dimethylformamide (DMF) | ~270 | High | Aprotic | Useful for dissolving many organic compounds. |
| Diethyl Ether | ~220 | Low | Non | Highly flammable; peroxides can form. |
*Approximate wavelength where absorbance = 1 in a 1 cm pathlength cell [42] [8].
Advanced Applications: Exploiting Solvatochromism
Beyond being a pitfall to avoid, solvatochromism can be harnessed as a powerful analytical tool to probe molecular structure and environment.
Distinguishing Structurally Similar Compounds
Recent research demonstrates that solvatochromic sensitivity can differentiate between compounds that are nearly identical, such as:
- Structural isomers (e.g., 1-propanol vs. 2-propanol) [61].
- Compounds differing by a single methylene group (e.g., glycine vs. alanine) [61].
- Proteins with minor conformational changes or point mutations [61].
The experimental protocol involves measuring the λmax of a solvatochromic probe dye in the presence of the different solutes. Even slight differences in the solute's ability to interact with the dye via polarity or hydrogen bonding can produce measurable shifts in the dye's absorption spectrum [61].
Determining Excited-State Dipole Moments
The magnitude of the solvatochromic shift can be quantified using linear solvent energy relationships (e.g., the Lippert-Mataga equation). By plotting the transition energy (in wavenumbers) against a solvent polarity function, the difference between the dipole moments of the ground and excited states (Δμ) can be calculated [62]. This provides deep insight into the charge redistribution that occurs upon photoexcitation.
Experimental Protocols
Protocol 1: Characterizing a Compound's Solvatochromic Behavior
Objective: To determine the λmax of a novel compound in various solvents and classify its solvatochromism. Materials: UV-Vis spectrophotometer, quartz cuvettes, analytical-grade solvents, analyte.
- Solution Preparation: Prepare stock solutions of the analyte at a low concentration (e.g., 10-100 µM) in a series of solvents spanning a wide polarity range (e.g., n-hexane, diethyl ether, ethyl acetate, chloroform, acetone, acetonitrile, methanol, water). Ensure solubility and avoid aggregation.
- Instrument Setup: Zero the spectrophotometer with a blank cuvette filled with the pure solvent for each measurement series.
- Spectral Acquisition: Place each sample solution in a quartz cuvette and acquire the full UV-Vis absorption spectrum from near the solvent's cutoff to 800 nm.
- Data Analysis: For each spectrum, identify the wavelength of maximum absorbance (λmax) for the key absorption band. Plot λmax versus a solvent polarity parameter (e.g., ET(30)) to visualize the solvatochromic trend.
Protocol 2: Using Solvatochromism to Detect Molecular Interactions
Objective: To use a solvatochromic dye as a probe for the local microenvironment. Materials: UV-Vis spectrophotometer, quartz cuvettes, a solvatochromic dye (e.g., 4-nitroaniline, Reichardt's dye), test solutes (e.g., isomers, proteins).
- Probe Solution: Prepare a standard solution of the solvatochromic dye in a universal solvent like water or a buffer.
- Titration: Add increasing concentrations of the test solute (e.g., a structural isomer) to the probe solution.
- Measurement: After each addition, measure the λmax of the dye's intramolecular charge-transfer band.
- Analysis: Plot the shift in λmax (or transition energy) against the concentration of the test solute. A differential shift between two similar solutes indicates a difference in their interaction with the dye's excited state [61].
The Scientist's Toolkit: Key Research Reagents & Materials
Table 3: Essential Materials for Solvatochromism Studies
| Item | Function & Importance |
|---|---|
| Quartz Cuvettes | Essential for UV-range measurements (<350 nm) as they are transparent down to ~190 nm, unlike glass or plastic [8]. |
| Spectroscopic-Grade Solvents | High-purity solvents with low UV absorbance ensure that measured signals originate from the analyte, not impurities. |
| Solvatochromic Probe Dyes | 4-Nitroanisole, N,N-Diethyl-4-nitroaniline, 4-Nitroaniline: A set of dyes with varying sensitivity to polarity and H-bonding, used to characterize microenvironments [61]. Reichardt's Dye: A well-known dye with extreme solvatochromism, used to define the ET(30) polarity scale. |
| Deuterium & Tungsten/Halogen Lamps | Standard light sources in UV-Vis instruments to cover the ultraviolet and visible ranges, respectively [8] [9]. |
| Monochromator | A critical optical component (often with a diffraction grating) that selects a specific, narrow wavelength of light to pass through the sample [8]. |
The following workflow summarizes the key decision points and steps for a robust solvent selection and analysis process.
Solvent selection is a critical, non-trivial step in UV-Vis spectroscopy that directly impacts data quality and interpretation. By grounding experimental design in the theory of electronic transitions, researchers can move from merely avoiding solvent-related errors to actively exploiting solvatochromic effects. Mastering the management of the solvent environment ensures accurate compound identification and quantification and unlocks advanced applications in probing molecular structure and microenvironments, a capability of immense value in fields from drug development to materials science.
The integrity of biological and chemical samples is a foundational prerequisite for successful analytical research, particularly in techniques like Ultraviolet-Visible (UV-Vis) spectroscopy where electronic transitions provide the fundamental measurement mechanism. Sample preparation methods directly influence the prevention of aggregation and degradation, which can significantly alter spectroscopic results by modifying the energy gaps between molecular orbitals. When samples aggregate, intermolecular interactions can shift absorption maxima, while degradation processes fragment molecules, potentially eliminating characteristic chromophores entirely. This guide establishes comprehensive protocols for maintaining sample integrity throughout preparation workflows, ensuring that observed UV-Vis spectra accurately reflect the true electronic structure of the molecules under investigation rather than preparation artifacts.
Core Theory: Electronic Transitions in UV-Vis Spectroscopy
UV-Vis spectroscopy measures the promotion of electrons from ground state to excited state molecular orbitals through photon absorption [13]. The energy required for this transition corresponds to specific wavelengths in the ultraviolet (200-400 nm) and visible (400-700 nm) regions of the electromagnetic spectrum according to the equation E = hc/λ, where E is energy, h is Planck's constant, c is the speed of light, and λ is wavelength [4].
Molecular Orbital Transitions
The most relevant transitions for organic molecules include:
- π → π* transitions: Occur in molecules with conjugated π systems, where electrons are promoted from bonding π orbitals to antibonding π* orbitals
- n → π* transitions: Involve the promotion of non-bonding electrons (lone pairs) to π* antibonding orbitals
- σ → σ* transitions: Require high energy and typically occur in the far-UV region (<200 nm) [13]
As conjugation increases in molecules, the energy gap (ΔE) between the Highest Occupied Molecular Orbital (HOMO) and Lowest Unoccupied Molecular Orbital (LUMO) decreases, resulting in longer wavelength absorption maxima (λmax) [4]. This relationship makes UV-Vis spectroscopy exceptionally sensitive to molecular changes that affect conjugation, including aggregation that alters π-orbital overlap or degradation that disrupts conjugated systems.
Major Degradation and Aggregation Pathways
Understanding the mechanisms of sample compromise enables researchers to implement targeted protective strategies throughout the preparation workflow.
Primary Degradation Mechanisms
Table 1: Major Degradation Pathways and Their Effects on Samples
| Mechanism | Causes | Impact on Samples | UV-Vis Spectral Impact |
|---|---|---|---|
| Oxidative Damage | Exposure to reactive oxygen species, heat, UV radiation [63] | Modifies nucleotide bases and amino acid side chains, creates strand breaks [63] | Altered λmax, reduced absorption intensity, new absorption bands |
| Hydrolytic Damage | Presence of water molecules, especially at extreme pH [63] | Breaks chemical bonds in backbone, causes depurination [63] | Decreased absorption at characteristic λmax due to chromophore loss |
| Enzymatic Degradation | Endogenous nucleases/proteases, microbial contamination [63] | Rapid breakdown of nucleic acids and proteins [63] | Progressive loss of absorption features over time |
| Mechanical Shearing | Overly aggressive homogenization, vortexing [63] | DNA fragmentation, protein denaturation [63] | Spectral broadening, reduced peak resolution |
Aggregation Mechanisms
Aggregation occurs through hydrophobic interactions, disulfide bonding, or electrostatic interactions, leading to multimolecular complexes that scatter light and create apparent absorption in UV-Vis spectra, potentially obscuring the true electronic transitions of monomeric species.
Sample Preparation Workflows
The following workflow diagrams and protocols outline standardized procedures for maintaining sample integrity across various sample types.
Generalized Sample Preparation Workflow
specialized workflow for challenging samples
Detailed Methodologies and Protocols
Standard Protein Extraction Protocol
Objective: Extract proteins while maintaining native state and preventing aggregation or degradation for UV-Vis analysis of chromophores.
Materials:
- Lysis buffer (50 mM Tris-HCl, 150 mM NaCl, 1% NP-40, pH 7.4)
- Protease inhibitor cocktail
- Refrigerated centrifuge
- Mechanical homogenizer (Bead Ruptor Elite or equivalent)
- Dialysis membrane or spin concentrators
Procedure:
- Sample Preservation: Flash-freeze tissue samples in liquid nitrogen immediately after collection. Store at -80°C if not processing immediately [63].
- Lysis Buffer Preparation: Add fresh protease inhibitors to chilled lysis buffer. Maintain at 4°C throughout procedure.
- Mechanical Disruption: Homogenize samples using optimized parameters (speed: 4-6 m/s, cycles: 3-5, duration: 30-60 seconds with cooling intervals) [63].
- Solubilization: Incubate homogenate with gentle agitation at 4°C for 30 minutes to extract membrane proteins.
- Clarification: Centrifuge at 12,000 × g for 15 minutes at 4°C. Collect supernatant carefully to avoid lipid layer.
- Buffer Exchange: Perform desalting using size exclusion chromatography or dialysis against appropriate spectroscopic buffer [64].
- Concentration Determination: Measure absorbance at 280 nm and appropriate secondary wavelengths to assess protein concentration and check for aggregation.
Troubleshooting:
- If aggregation is suspected (A340/A280 > 0.02), add stabilizing agents or adjust buffer conditions.
- For viscous samples, increase buffer volume or incorporate brief nuclease treatment.
DNA Extraction from Challenging Samples
Objective: Extract high molecular weight DNA with minimal fragmentation and degradation for UV-Vis analysis.
Materials:
- Extraction buffer (100 mM Tris-HCl, 10 mM EDTA, 1% SDS, pH 8.0)
- RNase A and Proteinase K
- Phenol:chloroform:isoamyl alcohol (25:24:1)
- Isopropanol and 70% ethanol
- Specialized bead tubes (ceramic or stainless steel)
Procedure:
- Sample Demineralization: For mineralized tissues, incubate in 0.5 M EDTA at 4°C for 24-48 hours with gentle agitation [63].
- Lysis Optimization: Use mechanical homogenization with controlled parameters (speed: 5 m/s, time: 2 × 45 seconds with 60-second cooling) [63].
- Enzymatic Digestion: Incubate with Proteinase K (100 μg/mL) at 55°C for 3 hours.
- Nuclease Treatment: Add RNase A (20 μg/mL) and incubate at 37°C for 30 minutes.
- Organic Extraction: Perform phenol:chloroform extraction twice, followed by chloroform extraction.
- Precipitation: Add 0.7 volumes isopropanol, mix gently, and spool DNA. Wash with 70% ethanol.
- Hydration: Resuspend in TE buffer (10 mM Tris-HCl, 0.1 mM EDTA, pH 8.0) at 4°C overnight.
Quality Assessment:
- Measure A260/A280 ratio (expect 1.8-2.0)
- Measure A260/A230 ratio (expect >2.0)
- Perform fragment analysis by agarose gel electrophoresis
Research Reagent Solutions
Table 2: Essential Reagents for Sample Preparation and Their Functions
| Reagent/Category | Specific Examples | Function in Sample Preparation | Considerations for UV-Vis |
|---|---|---|---|
| Lysis Buffers | Tris-HCl, HEPES, phosphate buffers | Maintain pH stability during extraction | Use buffers with low UV absorbance in spectral region of interest |
| Detergents | NP-40, Triton X-100, SDS, CHAPS | Solubilize membranes and prevent aggregation | Select detergents that don't interfere with chromophore absorbance |
| Reducing Agents | DTT, β-mercaptoethanol, TCEP | Prevent disulfide bond formation and aggregation | Fresh preparation required as many oxidize rapidly |
| Protease/Nuclease Inhibitors | PMSF, protease inhibitor cocktails, EDTA | Prevent enzymatic degradation during processing | EDTA can chelate metal cofactors in chromophores |
| Cryoprotectants | Glycerol, DMSO, sucrose | Stabilize samples during freezing | Can increase solution viscosity affecting path length |
| Solid-Phase Extraction Materials | C18, silica, polymer-based sorbents | Desalting and detergent removal [64] | Ensure no selective retention of analytes of interest |
| Preservation Solutions | RNAlater, AllProtect, formalin | Stabilize nucleic acids and proteins | Some fixatives may create chromophores that interfere |
Quality Control and Validation Methods
Robust quality control measures are essential to verify sample integrity before UV-Vis analysis. Both quantitative assessments and spectral validation provide critical information about preparation success.
Spectrophotometric Quality Assessment
Table 3: Quality Control Metrics for Different Sample Types
| Sample Type | Primary QC Metrics | Acceptance Criteria | Impact on UV-Vis Interpretation |
|---|---|---|---|
| Proteins | A260/A280 ratio, A340 scatter | A260/A280 < 0.6 (pure protein)A340/A280 < 0.02 | High scatter indicates aggregation affecting absorption measurements |
| Nucleic Acids | A260/A280, A260/A230, fragment analysis | A260/A280: 1.8-2.0A260/A230: >2.0 | Contaminants create background absorption; fragmentation alters hypochromicity |
| Small Molecules | Spectral shape, retention time | Comparison to reference standard | Aggregation can cause spectral shifts and reduced extinction coefficients |
Advanced Quality Control Techniques
For research requiring the highest reliability, these advanced methods provide additional validation:
- Size Exclusion Chromatography with Multi-Angle Light Scattering (SEC-MALS): Directly quantifies aggregation percentage and molecular mass distributions [64]
- Dynamic Light Scattering (DLS): Detects sub-visible aggregates and provides hydrodynamic radius measurements
- Capillary Electrophoresis: Assesses charge heterogeneity resulting from degradation or modification
- Liquid Chromatography-Mass Spectrometry (LC-MS): Identifies specific degradation products and modifications
Data Presentation and Documentation
Effective presentation of sample preparation methodology and quality control data ensures research reproducibility and proper interpretation of UV-Vis results.
Tabular Presentation of Sample Quality Metrics
Table 4: Example Sample Quality Control Report for UV-Vis Studies
| Sample ID | Preparation Method | [](]Protein] Concentration (μg/μL) | A260/A280 Ratio | A340/A280 Ratio | Aggregation Status | Recommended for UV-Vis |
|---|---|---|---|---|---|---|
| P-01 | Direct lysis, no filtration | 2.1 | 0.59 | 0.035 | Moderate aggregation | No - repeat with filtration |
| P-02 | Gentle lysis, 0.2μm filtered | 1.8 | 0.57 | 0.012 | Minimal aggregation | Yes - full spectral analysis |
| P-03 | Extended sonication | 2.4 | 0.62 | 0.081 | Significant aggregation | No - optimize lysis protocol |
| DNA-01 | Phenol-chloroform extraction | 0.15 | 1.89 | 2.31 | High molecular weight | Yes - ideal for spectroscopy |
Spectral Validation of Sample Integrity
Proper documentation includes representative spectra demonstrating sample quality:
- Include baseline-corrected spectra with clearly labeled absorption maxima
- Document spectral features indicative of degradation (increased baseline scattering, shifted λmax)
- Compare prepared samples against reference standards when available
- Report extinction coefficients calculated from concentration measurements
Implementing rigorous sample preparation protocols that prevent aggregation and degradation is essential for obtaining meaningful UV-Vis spectroscopic data. By understanding the fundamental electronic transitions measured by UV-Vis spectroscopy, researchers can better appreciate how sample integrity directly influences spectral outcomes. The methodologies presented here provide a framework for maintaining sample quality across diverse applications, ensuring that observed spectral features genuinely represent the molecular electronic structures under investigation rather than preparation artifacts. As UV-Vis methodologies continue to advance in sensitivity and application, the principles of careful sample handling and thorough quality control remain constant requirements for generating reliable, reproducible spectroscopic data.
Ultraviolet-Visible (UV-Vis) spectroscopy fundamentally probes the electronic structure of molecules by measuring their absorption of light, which promotes electrons from ground states to higher energy levels [8]. The accuracy of this technique in characterizing π→π*, n→π*, and charge-transfer transitions depends critically on the integrity of the instrumental signal [65]. Artifacts such as stray light, improper bandwidth, and suboptimal cuvette selection directly distort the measured absorbance, leading to deviations from the Beer-Lambert law and erroneous conclusions about molecular concentration or identity [66] [67]. This guide details the sources, consequences, and mitigation strategies for these key instrumental artifacts, providing a framework for obtaining reliable spectroscopic data in research and drug development.
Theoretical Definition and Impact on Electronic Transitions
Stray light is defined as any light reaching the detector that lies outside the nominal wavelength band selected by the monochromator [66]. It arises from scatter, diffraction, or internal reflections within the optical path [66] [68]. This polychromatic component introduces a significant error in measured absorption because it constitutes light that has not engaged with the electronic transitions of the sample. The effect is most pronounced at high absorbance values where the transmitted light intensity is low. The stray light component (Istray) adds to the true transmitted light (I), leading to a measured absorbance (Ameas) that is lower than the true absorbance (Atrue), causing a negative deviation from the Beer-Lambert law [66]: Ameas = -log10( (I + Istray) / I0 ) This effect is particularly critical in the UV region, where many chromophores relevant to drug development (e.g., aromatic amino acids, nucleic acids) absorb strongly, and where the energy throughput of the instrument is naturally lower [66] [8].
Experimental Protocols for Stray Light Monitoring
Regular validation is essential. The following standardized procedures are used to quantify stray light:
ASTM Procedure: This method uses cut-off filters that strongly absorb light below a specific wavelength but transmit at longer wavelengths. Any light detected below this cut-off is identified as stray light [66].
- Materials: Sealed cuvettes containing (1) 10 g/L Sodium Iodide for testing at 220 nm, (2) 50 g/L Sodium Nitrite for testing at 340 nm and 370 nm [66].
- Method: The transmittance of the solution is measured at the specified wavelength. A transmittance reading significantly above 0% indicates the presence of stray light [66].
Pharmacopoeial Procedure: The European Pharmacopoeia recommends using a 12 g/L potassium chloride solution and measuring its absorbance at 198 nm. The absorbance reading should be greater than 2.0 Absorbance Units (AU) [66].
Table 1: Standard Solutions for Stray Light Testing
| Solution | Concentration | Test Wavelength | Acceptance Criterion |
|---|---|---|---|
| Sodium Iodide | 10 g/L | 220 nm | Low Transmittance (%) |
| Sodium Nitrite | 50 g/L | 340 nm & 370 nm | Low Transmittance (%) |
| Potassium Chloride | 12 g/L | 198 nm | Absorbance > 2.0 AU |
Visualizing Stray Light's Effect on Data Integrity
The following diagram illustrates how stray light originates within a spectrophotometer and its subsequent effect on analytical data, showing the deviation from the Beer-Lambert law.
Diagram 1: Origin and effect of stray light on spectral data.
Spectral Bandwidth: Resolving Electronic Transitions
The Principle of Bandwidth
The spectral bandwidth (SBW) of a spectrometer is the width of the wavelength interval exiting the monochromator. It is determined by the physical width of the entrance and exit slits and the dispersion of the grating [68]. It is typically defined as the Full Width at Half Maximum (FWHM) of the bandpass [68]. The SBW dictates the instrument's ability to resolve fine spectral features, such as the sharp vibronic transitions found in many organic molecules.
Bandwidth-Induced Errors in Quantitative Analysis
Using an SBW that is too wide can lead to two types of errors:
- Decreased Apparent Absorbance: For samples with sharp absorption peaks, a wide SBW delivers polychromatic light to the sample. Since the molar absorptivity (ε) varies across the bandpass, the measured absorbance is an average that is less than the peak absorbance. This violates the fundamental assumption of the Beer-Lambert law for monochromatic light [67].
- Loss of Spectral Resolution: Closely spaced peaks may not be distinguished as separate entities if the SBW is larger than the separation between them [68]. This is critical for identifying specific chromophores in a complex mixture, a common scenario in drug development.
Protocol for Verifying Spectral Resolution
A practical method to verify the instrument's resolution is to measure a substance with known, sharp absorption peaks.
- Recommended Standard: A holmium oxide filter or holmium in solution (perchlorate or nitrate) displays sharp absorption peaks at well-defined wavelengths (e.g., 360.8 nm, 418.5 nm, 453.2 nm, 536.4 nm) [67].
- Method: Scan the standard and measure the FWHM of an isolated peak to confirm the instrumental SBW. Alternatively, check that the instrument can resolve the distinct doublet at ~536 nm and ~536.4 nm in a holmium filter. Failure to resolve these features indicates the effective bandwidth is too large for high-resolution work [67].
Cuvette Selection: The Sample-Optics Interface
The cuvette is a critical optical component, not just a container. Its material, path length, and condition directly interact with the light beam and can introduce significant artifacts.
Cuvette Material and Wavelength Transmission
The choice of material dictates the usable wavelength range of an experiment, as different materials have distinct UV-Vis transmission cut-offs.
Table 2: Cuvette Material Properties and Applications
| Material | Transmission Range | Transmission Rate (Example) | Primary Application | Chemical Resistance |
|---|---|---|---|---|
| Optical Glass | 340 – 2,500 nm [69] [70] | >80% at 350 nm [69] | Visible light analyses [71] | Good, avoid strong alkali [71] |
| ES/UV Quartz | 190 – 2,500 nm [69] [70] | >83% at 220 nm [69] | Deep-UV (proteins, DNA) [71] | Excellent (acids, bases, solvents) [71] [70] |
| IR Quartz | 220 – 3,500 nm [69] | ~88% at 2730 nm [69] | Extended UV-Vis-NIR studies | Excellent [69] |
| Plastic (PS/PMMA) | 380 – 780 nm [69] [70] | ~80% at 400 nm [69] | Disposable visible assays [69] | Poor (organic solvents) [69] |
Using a glass or plastic cuvette for a deep-UV measurement (e.g., at 260 nm for DNA) will result in significant light absorption by the cuvette itself, manifesting as excessive noise, low light throughput, and effectively, stray light [8] [69].
Path Length and Its Dimensional Tolerance
The path length (L) is a direct variable in the Beer-Lambert law (A = εcL). The industry standard for path length tolerance is typically ±0.05 mm for a standard 10 mm cuvette [69] [70]. This small variation can introduce a systematic error of up to 0.5% in concentration measurements. Furthermore, imperfections like cuvette tilt or wedge (non-parallel windows) can alter the effective path length and cause beam deviation, leading to signal loss and photometric error [67].
Cuvette Handling and Cleaning Best Practices
Improper handling is a major source of artifacts.
- Cleaning: Rinse immediately after use with a compatible solvent. Use lint-free swabs to avoid scratches. Avoid ultrasonic baths for fused quartz cuettes, as vibrations can damage optical surfaces [71].
- Handling: Always wear gloves to prevent fingerprints, which absorb significantly in the 270-300 nm range [71]. Store cuvettes clean and dry to prevent microbial growth or etching [71].
- Inspection: Visually inspect cuvettes for scratches, cracks, or degraded coatings before use. Damaged cuvettes are a primary source of light scatter and stray light [68].
The Scientist's Toolkit: Essential Research Reagents and Materials
The following table lists key materials and standards required for the validation and mitigation protocols discussed in this guide.
Table 3: Essential Research Reagents and Materials for UV-Vis Artifact Management
| Item | Function / Application | Key Specifications |
|---|---|---|
| Stray Light Calibration Standards | Validation of spectrophotometer stray light performance [66] [72] | NIST-traceable solutions (KCl, NaI, NaNOâ‚‚) or solid filters [66] [72] |
| Holmium Oxide Filter/Solution | Verification of wavelength accuracy and spectral resolution/bandwidth [67] | Certified sharp absorption peaks at defined wavelengths (e.g., 360.8, 453.2, 536.4 nm) |
| Quartz Cuvettes (ES Grade) | Sample holder for UV-Vis measurements, especially in deep-UV range [71] [69] | Transmission range: 190-2500 nm; Path length: 10 mm (±0.05 mm tolerance) |
| Neutral Density Filters | Checking photometric linearity of the instrument [67] [72] | Certified absorbance values at specific wavelengths |
| Lint-Free Wipes/Swabs | Safe cleaning of quartz and glass cuvettes without scratching [71] | Microfiber or foam-tip material |
A deep understanding of electronic transitions in UV-Vis spectroscopy must be paired with a rigorous approach to instrumental validation. Stray light, improper bandwidth, and cuvette-related artifacts are not minor technicalities but fundamental sources of error that can compromise quantitative analysis and structural characterization. By implementing the systematic monitoring and calibration procedures outlined here—regular stray light checks, bandwidth verification, and prudent cuvette selection and handling—researchers and drug development professionals can ensure the integrity of their spectroscopic data, leading to more reliable and reproducible scientific outcomes.
Ultraviolet-Visible (UV-Vis) spectroscopy functions by measuring the absorption of light in the ultraviolet and visible regions of the electromagnetic spectrum, typically between 200 and 800 nm [2] [3]. When a molecule absorbs light, an electron is promoted from a ground state orbital to a higher energy, excited state orbital [2] [4]. The energy of the absorbed photon must exactly match the energy difference (( \Delta E )) between these two electronic states, a relationship defined by the equation ( \Delta E = h\nu ), where ( h ) is Planck's constant and ( \nu ) is the frequency of the light [4] [11]. The probability and energy of these transitions are influenced by the molecule's chemical structure and its immediate environment. Changes in these factors lead to measurable shifts in the absorption spectrum, categorized as bathochromic, hypsochromic, hyperchromic, and hypochromic effects [73] [74] [75]. These shifts provide critical insights for researchers in drug development, enabling them to probe molecular interactions, conformational changes, and the stability of compounds under various conditions.
Theoretical Foundation of Electronic Transitions
Types of Electronic Transitions
The primary electronic transitions observed in UV-Vis spectroscopy involve the excitation of electrons in molecular orbitals. For organic molecules, the most significant transitions involve ( \pi ), ( \sigma ), and ( n ) (non-bonding) electrons [2] [3]. The common types of electronic transitions are:
- ( \pi \rightarrow \pi^* ) Transitions: These occur in molecules with unsaturated centers, such as alkenes, alkynes, and carbonyl compounds, where an electron is excited from a bonding ( \pi ) orbital to an antibonding ( \pi^* ) orbital [2]. They are typically strong transitions with high molar absorptivities (ε between 1,000 and 10,000 L molâ»Â¹ cmâ»Â¹) and are sensitive to conjugation [2] [5].
- ( n \rightarrow \pi^* ) Transitions: These involve the excitation of a non-bonding electron (e.g., on oxygen or nitrogen) to a ( \pi^* ) orbital [2]. These transitions are of lower intensity (ε from 10 to 100 L molâ»Â¹ cmâ»Â¹) and are common in carbonyl compounds [2] [5]. They are highly sensitive to solvent effects, often shifting to shorter wavelengths (blue shift) with increasing solvent polarity [2].
- ( \sigma \rightarrow \sigma^* ) Transitions: These require high energy, corresponding to light in the far-UV region (e.g., λ~125 nm for methane), and are thus rarely observed in standard UV-Vis spectra [2] [4].
- ( n \rightarrow \sigma^* ) Transitions: These occur in saturated compounds with lone pairs, absorbing light in the range of 150-250 nm [2].
- Charge-Transfer Transitions: These involve the transfer of an electron from a donor to an acceptor moiety within a molecule or complex, resulting in very intense absorption bands (ε > 10,000 L molâ»Â¹ cmâ»Â¹) [2] [74].
The Role of Chromophores and Auxochromes
A chromophore is the part of a molecule responsible for its absorption of UV or visible light, typically a region with a conjugated ( \pi )-electron system or a functional group with heteroatoms possessing non-bonding electrons [74] [5] [76]. Examples include C=C, C=O, N=N, and nitro groups [74] [75]. The presence of a chromophore is a prerequisite for absorption.
An auxochrome is a functional group attached to the chromophore that itself does not absorb significantly in the UV-Vis region but modifies the chromophore's absorption by interacting with its ( \pi )-system [75] [76]. Common auxochromes include -OH, -NHâ‚‚, and -OR [75]. They typically induce a bathochromic (red) shift and/or a hyperchromic effect (increased intensity) by extending the conjugation or through resonance effects [75] [76].
Figure 1: Fundamental process of electronic transitions and the origin of spectral shifts.
Classification and Mechanism of Spectral Shifts
Spectral shifts are categorized based on the direction of the wavelength shift or the change in absorption intensity. The following table provides a definitive summary of these effects.
Table 1: Classification of Spectral Shifts in UV-Vis Spectroscopy
| Shift Type | Alternative Name | Direction of Change | Molecular Origin |
|---|---|---|---|
| Bathochromic | Red Shift | Shift to Longer Wavelength (Lower Energy) [73] [74] | Increased conjugation, solvent polarity change (positive solvatochromism), or auxochrome introduction [73] [75] [76]. |
| Hypsochromic | Blue Shift | Shift to Shorter Wavelength (Higher Energy) [73] [74] | Removal of conjugation, change in solvent polarity (negative solvatochromism), or protonation of a group [73] [75]. |
| Hyperchromic | - | Increase in Molar Absorptivity (ε) [73] [75] | Introduction of an auxochrome or structural change that increases the probability of the electronic transition [73] [75]. |
| Hypochromic | - | Decrease in Molar Absorptivity (ε) [73] [75] | Structural deformation or introduction of a group that decreases the transition probability [73] [75]. |
Bathochromic Shift (Red Shift)
A bathochromic shift describes the movement of an absorption band to a longer wavelength, indicating a reduced energy gap (( \Delta E )) between the ground and excited states [73]. A classic example is the increase in ( \lambda_{max} ) with extended conjugation in polyenes: ethene (170 nm), butadiene (217 nm), and hexatriene (258 nm) [4]. This occurs because conjugation lengthens the ( \pi )-system, raising the energy of the Highest Occupied Molecular Orbital (HOMO) and lowering the energy of the Lowest Unoccupied Molecular Orbital (LUMO), thereby shrinking the HOMO-LUMO gap [4] [76]. In drug development, this effect can signal the intercalation of a small molecule into DNA, as the planar aromatic ring system of the drug interacts with the base pairs, effectively extending the conjugated system.
Hypsochromic Shift (Blue Shift)
A hypsochromic shift is the movement of an absorption maximum to a shorter wavelength, indicating an increased energy gap (( \Delta E )) [73]. This can occur when the conjugation in a molecule is disrupted. For instance, aniline shows a ( \lambda_{max} ) at 280 nm due to conjugation between the lone pair on nitrogen and the benzene ring. In acidic conditions, protonation of the -NH₂ group to -NH₃⺠removes this lone pair from conjugation, resulting in a hypsochromic shift and a spectrum resembling that of benzene (~255 nm) [75]. This principle is used to study the ionization states of drug molecules.
Hyperchromic Effect
The hyperchromic effect is an increase in the intensity of absorption (molar absorptivity, ε) [73] [75]. This occurs when a structural change increases the probability of the electronic transition. The attachment of an auxochrome like -OH or -NH₂ to a chromophore often produces both a bathochromic and a hyperchromic shift [75]. A key biochemical example is the hyperchromic shift observed when double-stranded DNA denatures. The stacked base pairs in native DNA exhibit suppressed absorption due to ( \pi )-stacking interactions. Denaturation separates the strands, freeing the bases and leading to a significant increase in absorbance at around 260 nm [75].
Hypochromic Effect
The hypochromic effect is a decrease in the intensity of absorption (molar absorptivity, ε) [73] [75]. It is typically caused by structural changes that deform the chromophore or introduce steric hindrance, reducing the efficiency of the electronic transition. In pharmaceutical sciences, the formation of a complex between a drug and a biomolecule might lead to a hypochromic effect if the interaction restricts the vibrational or rotational freedom of the chromophore or alters its local electronic environment.
Table 2: Quantitative Impact of Conjugation on Absorption Maxima (π→π Transitions)*
| Compound | Number of Conjugated Double Bonds | ( \lambda_{max} ) (nm) | Observation |
|---|---|---|---|
| Ethene | 1 | ~170 [4] | Colorless (Absorbs in far UV) |
| 1,3-Butadiene | 2 | ~217 [4] | Colorless (Absorbs in UV) |
| 1,3,5-Hexatriene | 3 | ~258 [4] | Colorless (Absorbs in UV) |
| Lycopene | 11 | ~450-500 [76] | Red (Absorbs in visible region) |
Solvatochromism: A Key Environmental Effect
Solvatochromism is the change in the color (absorption spectrum) of a substance due to a change in the polarity of the solvent [73]. It is a critical phenomenon for drug developers, as a drug's environment in the body can vary from aqueous (blood) to non-polar (lipid membranes).
- Positive Solvatochromism: A bathochromic (red) shift occurs with increasing solvent polarity. This indicates that the excited state is more polar and thus stabilized more by polar solvents than the ground state, reducing the energy gap (( \Delta E )) [73].
- Negative Solvatochromism: A hypsochromic (blue) shift occurs with increasing solvent polarity. This indicates that the ground state is more polar than the excited state. The ground state is stabilized more in polar solvents, increasing the energy gap (( \Delta E )) [73].
The direction of the shift provides insight into the relative polarity of a molecule's ground versus excited state, which is valuable for understanding its photophysical properties.
Figure 2: Mechanism of solvatochromism, showing how solvent polarity differentially stabilizes electronic states to cause spectral shifts.
Experimental Protocols and Methodologies
Protocol 1: Observing Solvatochromic Shifts
Objective: To demonstrate the effect of solvent polarity on the absorption spectrum of a solvatochromic dye.
Sample Preparation:
- Select a solvatochromic compound (e.g., a ketone with n→π* transition or a charge-transfer complex).
- Prepare stock solutions of the compound in a series of solvents of increasing polarity (e.g., cyclohexane, diethyl ether, dichloromethane, ethanol, water). Ensure the compound is soluble in all chosen solvents.
- Dilute each stock solution to an appropriate concentration, typically yielding an absorbance between 0.2 and 1.0 AU for the peak of interest, using the same solvent for dilution. Use matched quartz cuvettes (path length, typically 1 cm).
Instrumental Setup:
- Use a double-beam UV-Vis spectrophotometer to automatically subtract solvent background.
- Set instrument parameters: wavelength range (e.g., 200-600 nm), scan speed (medium), and spectral bandwidth (as narrow as possible for highest resolution, often 1-2 nm) [3].
- Allow the lamp and instrument to warm up for the recommended time to ensure stability.
Data Acquisition:
- Blank the spectrometer with a cuvette containing only the pure solvent.
- Place the cuvette containing the sample solution in the sample holder.
- Run the scan and record the spectrum.
- Repeat the blanking and scanning procedure for each solvent in the series.
- Ensure all spectra are saved with clear labels.
Data Analysis:
- Identify the wavelength of maximum absorption (( \lambda_{max} )) for the key peak in each solvent.
- Plot ( \lambda_{max} ) versus a quantitative measure of solvent polarity (e.g., dielectric constant, ET(30) parameter).
- A positive slope indicates positive solvatochromism; a negative slope indicates negative solvatochromism.
Protocol 2: Quantifying the Hyperchromic Effect in DNA Denaturation
Objective: To measure the increase in absorbance at 260 nm as double-stranded DNA denatures into single strands.
Sample Preparation:
- Prepare a buffered aqueous solution of double-stranded DNA (e.g., calf thymus DNA). The absorbance at 260 nm should be about 0.5-1.0 for the native state to remain within the linear range of the detector after denaturation [3].
- Aliquot the DNA solution into several vials for different temperature points or denaturant concentrations.
Instrumental Setup:
- Use a UV-Vis spectrophotometer equipped with a temperature-controlled cell holder.
- Set the instrument to monitor absorbance at a fixed wavelength (260 nm).
Data Acquisition (Thermal Denaturation):
- Place the DNA sample in a thermally jacketed cuvette.
- Increase the temperature gradually (e.g., 1-2 °C per minute) while continuously recording the absorbance at 260 nm.
- Continue heating until the absorbance value plateaus, indicating complete denaturation.
Data Analysis:
- Calculate the hyperchromic effect as ( (Af - Ai)/Ai \times 100\% ), where ( Ai ) is the initial absorbance of native DNA and ( A_f ) is the final absorbance of denatured DNA.
- Plot absorbance versus temperature to generate a melting curve. The melting temperature (( T_m )) is the midpoint of this transition and provides information about the DNA's stability.
Table 3: Essential Research Reagent Solutions for Spectral Shift Studies
| Reagent / Material | Function / Application | Key Considerations |
|---|---|---|
| Spectrophotometric Grade Solvents | Used to prepare sample solutions; minimizes interfering absorbance [3]. | Must be transparent in the spectral region of interest. Common: water, ethanol, hexane, acetonitrile [3]. |
| Quartz Cuvettes | Hold liquid samples in the light path of the spectrometer. | Required for UV range (<350 nm); path length (e.g., 1 cm) must be known and consistent for concentration calculations [3]. |
| Buffers (e.g., Phosphate, Tris) | Maintain constant pH for studies of pH-sensitive chromophores (halochromism) [76]. | Buffer should not absorb in the studied region. |
| Standard Chromophores | Used for instrument calibration and validation of spectral shifts. | Examples: β-carotene (λ~452 nm) [76], potassium dichromate (for UV wavelength accuracy checks) [3]. |
| Denaturants (e.g., Urea, Heat) | Induce structural changes to study hyper/hypochromic effects. | Used in studies of biomolecules like DNA/RNA and proteins [75]. |
The interpretation of bathochromic, hypsochromic, hyperchromic, and hypochromic shifts is fundamental to extracting meaningful information from UV-Vis spectroscopy. These shifts are not merely spectral curiosities; they are direct reporters on the electronic structure, conformation, and microenvironment of molecules. For researchers and drug development professionals, a deep understanding of these effects enables the investigation of critical phenomena such as molecular binding interactions, DNA denaturation, protein-ligand complex formation, and the stability of compounds in different solvents and pH conditions. By systematically applying the protocols and principles outlined in this guide, scientists can leverage spectral shifts as powerful, quantitative tools for analytical characterization and mechanistic studies in pharmaceutical development.
Ultraviolet-visible (UV-Vis) spectroscopy serves as a fundamental tool for investigating the electronic structure of molecules by measuring their absorption of light in the ultraviolet and visible regions of the electromagnetic spectrum (typically 190–800 nm) [8] [9]. When photons in this energy range interact with matter, electrons in chromophores—light-absorbing molecular components—undergo transitions from ground states to excited states [4] [9]. The energy required for these promotions corresponds precisely to the difference between molecular orbitals (π-π, n-π, etc.), with conjugated systems exhibiting particularly distinctive absorption patterns due to their delocalized π-electrons [4] [77].
The interpretation of experimental UV-Vis spectra presents significant challenges, especially for complex conjugated molecules where overlapping transitions, vibronic coupling, and solvent effects obscure individual electronic transitions [78]. Traditional spectral fitting approaches relying on symmetric Gaussian or Lorentzian functions often fail to accurately represent the inherently asymmetric band shapes of electronic transitions, particularly at lower temperatures [78]. This technical guide introduces the Pekarian function (PF) fit as an advanced analytical framework that overcomes these limitations, enabling researchers to extract maximum quantitative information from experimental spectra while providing a more physically meaningful connection to underlying electronic processes.
Theoretical Foundation of the Pekarian Function
Historical Development and Physical Basis
The Pekarian function originates from theoretical work conducted 75 years ago by Huang, Rhys, and Pekar to describe the shape of absorption bands associated with F-centers in crystals [78]. These theories incorporated two fundamental simplifications: (a) the lattice could be approximated as a dielectric continuum, and (b) the effect of the F-center could be considered as that of a static charge distribution [78]. Krivoglaz and Pekar later concluded that this analysis of spectral shapes in crystals could be extended to liquids and even gases involving large polyatomic molecules, essentially treating each molecule as a "small crystal" [78].
The modern adaptation of this approach for conjugated molecules in solution incorporates the Huang-Rhys factor S, which represents the mean number of phonons (or vibrational quanta) accompanying the optical transition [78]. In the context of molecular systems in solution, this translates to vibrational modes interacting with the electronic transition. The PF approach assumes a single dominant vibrational mode of wavenumber Ω that interacts with the optical transition, though the method can be extended to multiple modes for gas-phase spectra with very narrow bands [78].
Mathematical Formulation
The modified Pekarian function for analyzing experimental UV-Vis spectra employs distinct expressions for absorption and fluorescence, accounting for the progression of vibronic transitions [78]. For absorption spectra (PFa), the function takes the form:
$$PFa(ν) = \sum{k=0}^{n} \frac{S^k e^{-S}}{k!} \times G(1, νk, σ_0)$$
where $νk = ν0 + kΩ + δ \times k(k-1)$ for absorption, and for fluorescence spectra (PFf):
$$PFf(ν) = \sum{k=0}^{n} \frac{S^k e^{-S}}{k!} \times G(1, νk, σ_0)$$
where $νk = ν0 - kΩ + δ \times k(k-1)$ for fluorescence [78].
In practice, the summation from k = 0–8 proves sufficient for most cases, with further increases showing negligible improvement in fitting quality [78]. The function optimizes five key parameters that define the band shape: S (Huang-Rhys factor), ν₀ (central wavenumber for the 0-0 transition), Ω (vibrational wavenumber), σ₀ (Gaussian broadening parameter), and δ (global correction compensating for contributions from other vibrational modes) [78].
Table 1: Definition of Parameters in the Modified Pekarian Function
| Parameter | Physical Significance | Typical Units |
|---|---|---|
| S | Huang-Rhys factor; represents the mean number of vibrational quanta accompanying the optical transition | Dimensionless |
| ν₀ | Central wavenumber for the 0-0 transition | cmâ»Â¹ |
| Ω | Effective vibrational wavenumber of the dominant mode interacting with the electronic transition | cmâ»Â¹ |
| σ₀ | Gaussian broadening parameter, characteristic for each spectrum | cmâ»Â¹ |
| δ | Global correction compensating contributions from other vibrational modes | cmâ»Â¹ |
For transitions involving charge transfer, Marcus proposed expressions analogous to the Pekarian function where these parameters manifest as specific functions of the charge transfer model [78]. The weighted average transition energy 〈νꜢₑ*〉 can be calculated from the fitted parameters using the relation:
$$〈ν{ge*}〉 = ν0 + Ω × S$$
This value proves particularly valuable for comparison with theoretical excitation energies calculated using time-dependent density functional theory (TD-DFT) [78].
Experimental Implementation and Protocols
Instrumentation and Measurement Considerations
Proper collection of UV-Vis spectral data requires understanding fundamental instrumentation principles. A UV-Vis spectrophotometer consists of four key components: a light source (typically xenon, deuterium, or tungsten/halogen lamps), a wavelength selector (monochromator or filters), a sample container (cuvette or cell), and a detector (photomultiplier tube, photodiodes, or CCD) [8] [9].
For reliable PF analysis, several measurement considerations prove critical:
Solvent Selection: Choose solvents that don't absorb significantly in the spectral region of interest. Remember that plastic cuvettes are inappropriate for UV absorption studies, and even glass absorbs most UVB and UVC radiation—quartz sample holders are required for UV examination [8].
Sample Concentration: Optimize concentration to maintain absorbance values below 1.0 within the dynamic range of the instrument, as absorbance exceeding this value means less than 10% of incoming light reaches the detector, compromising quantification reliability [8].
Temperature Control: Implement temperature stabilization, as electronic properties of conjugated organic derivatives and their dipole moments are highly temperature-dependent [78].
Baseline Correction: Exercise caution with baseline correction, as it may distort absorption bands. For the rubrene analysis, the researchers specifically avoided baseline correction prior to fitting to prevent band distortion [78].
Spectral Fitting Workflow
The process of implementing Pekarian function fits follows a systematic workflow that transforms raw spectral data into physically meaningful parameters.
Diagram 1: Pekarian Function Fitting Workflow
Case Study: Rubrene Analysis
The analysis of rubrene (5,6,11,12-tetraphenyltetracene) in toluene solution illustrates a practical application of the PF fitting approach [78]. The experimental absorption spectra collected at different temperatures show systematic intensity increases and band narrowing with decreasing temperature, accompanied by a bathochromic shift of the overall band [78].
For the main absorption band of rubrene at 20°C, the optimized parameters were: S = 0.87, ν₀ = 18941 cmâ»Â¹, Ω = 1353.7 cmâ»Â¹, σ₀ = 448.3 cmâ»Â¹, and δ = 15.1 [78]. A minor second component was required to separate the absorption tail from higher-energy transitions but lacked physical significance [78].
Temperature-dependent studies revealed that S remained practically constant at 0.87, while Ω exhibited weak temperature dependence (1352–1365 cmâ»Â¹ across 5–90°C) [78]. More pronounced temperature effects were observed for σ₀ (increasing from 437 to 500 with rising temperature) and δ (decreasing from 20 to 0 over the same range) [78]. The parameter ν₀ increased from 18923 to 19030 cmâ»Â¹ across this temperature range, with linear regression yielding excellent correlation (r² = 1) and extrapolation to absolute zero giving 18573 cmâ»Â¹ [78].
Table 2: Temperature Dependence of PF Parameters for Rubrene in Toluene
| Temperature (°C) | S | ν₀ (cmâ»Â¹) | Ω (cmâ»Â¹) | σ₀ (cmâ»Â¹) | δ |
|---|---|---|---|---|---|
| 5 | 0.87 | 18923 | 1352 | 437 | 20 |
| 20 | 0.87 | 18941 | 1353.7 | 448.3 | 15.1 |
| 90 | 0.87 | 19030 | 1365 | 500 | 0 |
Computational Integration and Advanced Applications
Correlation with Quantum Mechanical Calculations
The PF fitting approach achieves its full potential when integrated with computational quantum chemistry methods. Time-dependent density functional theory (TD-DFT) calculations provide crucial independent estimates of excitation energies that guide the determination of how many PF components are needed to fit a given experimental spectrum [78]. The weighted average 〈νꜢₑ*〉 derived from PF parameters enables direct comparison with theoretically calculated transition energies, creating a powerful feedback loop between experimental spectroscopy and computational chemistry [78].
For complex molecular systems, more advanced computational approaches such as QM/MM (quantum mechanics/molecular mechanics) methods may be employed, where a local region of interest is treated with quantum chemistry calculations and the surrounding environment with molecular mechanics force fields [79]. These hybrid approaches make it feasible to calculate molecular properties of complex systems, including spectra, with reasonable computational cost [79].
Handling Complex Spectral Scenarios
Real-world conjugated molecules often exhibit complex spectral features requiring multiple PF components:
Overlapping Bands: Spectra with partially or fully overlapping bands require several PF components for adequate fitting, each with its own set of parameters [78]. The rubrene analysis exemplifies this approach, where a second minor component was necessary to account for the absorption tail from higher-energy transitions [78].
Vibronic Progressions: For molecules with strong electron-phonon coupling, the Pekarian function naturally captures the Poisson progression of vibronic transitions through the summation over k values, properly weighting each vibronic band according to the Huang-Rhys factor [78].
Solvatochromic Effects: Moderate to strong solvatochromic shifts, often overlooked in spectral analysis, can be systematically investigated through PF analysis of spectra collected in different solvents [78].
Essential Research Tools and Reagents
Successful implementation of advanced spectral analysis using Pekarian function fits requires specific computational tools and laboratory resources.
Table 3: Essential Research Toolkit for PF Spectral Analysis
| Tool/Category | Specific Examples | Function/Purpose |
|---|---|---|
| Spectroscopy Software | PeakFit, Origin (with user-defined functions) | Commercial software platforms for implementing PF fits with customized function definitions [78] |
| Programming Tools | Python with custom PekarFit script | Open-source alternative for PF fitting; offers detailed outputs and deeper insight into fitting process [78] |
| Computational Chemistry Software | Gaussian, Q-Chem, DFTB+, TeraChem | Quantum chemistry programs for TD-DFT calculations to guide PF component selection [78] [79] |
| Sample Containment | Quartz cuvettes (for UV studies) | Material transparent to UV light; essential for measurements below 350 nm [8] |
| Solvent Systems | Toluene, acetonitrile, dimethyl sulfoxide | Appropriate solvents with minimal absorption in spectral region of interest [78] [8] |
| Temperature Control | Peltier-cooled cuvette holders | Temperature stabilization accessories for studying thermal effects on electronic transitions [78] |
The Pekarian function fit represents a sophisticated advancement beyond conventional symmetric function approaches for analyzing UV-Vis spectra of conjugated molecules. By incorporating physically meaningful parameters that connect directly to fundamental aspects of electronic transitions and vibronic coupling, the PF approach enables researchers to extract maximum quantitative information from experimental spectra. The method's compatibility with modern computational chemistry techniques creates a powerful synergy between experiment and theory, particularly valuable for complex systems such as organic donor-acceptor substituted dyes with semiconducting properties and nonlinear optical responses.
As UV-Vis spectroscopy continues to find applications across pharmaceuticals, materials science, and chemical research, advanced analytical frameworks like the Pekarian function fit will play an increasingly important role in unlocking the rich information content hidden within electronic absorption and emission spectra. The systematic protocol outlined in this technical guide provides researchers with a comprehensive roadmap for implementing this powerful approach in their investigations of conjugated molecular systems.
Beyond UV-Vis: Validating and Correlating Data with Complementary Analytical Techniques
Ultraviolet-Visible (UV-Vis) spectroscopy remains a cornerstone technique in the modern analytical laboratory, providing critical insights into molecular structure and concentration across diverse scientific fields. This absorption spectroscopy technique measures the amount of discrete wavelengths of UV or visible light that are absorbed by or transmitted through a sample in comparison to a reference or blank sample [8]. The fundamental principle governing this interaction is the promotion of electrons from their ground state to higher energy states when molecules absorb specific amounts of energy corresponding to light in the 190-800 nm range [8] [80].
In the context of contemporary analytical science, UV-Vis spectroscopy maintains its relevance through an evolving combination of technical robustness and adaptability to new challenges. The global UV-Vis spectroscopy market, valued at approximately USD 21.52 billion in 2025 and projected to reach USD 27.62 billion by 2030, demonstrates the technique's continued importance amid advancing analytical technologies [81]. This growth is particularly driven by sturdy demand from pharmaceutical continuous-manufacturing applications, where inline UV sensors enable real-time tracking of critical quality attributes [81]. This article assesses the role of UV-Vis spectroscopy by examining its fundamental theoretical principles, core strengths, inherent limitations, methodological protocols, and evolving applications within the modern analytical toolkit.
Theoretical Foundation: Electronic Transitions
At its core, UV-Vis spectroscopy probes the electronic structure of molecules through the mechanism of electron excitation. When atoms or molecules absorb energy from UV or visible light, electrons are promoted from their ground state to an excited state [2]. The specific energy required for these transitions corresponds to particular wavelengths of light, creating the characteristic absorption spectra that provide both qualitative and quantitative information about the sample [13].
Types of Electronic Transitions
The electronic transitions observable in the UV-Vis range fall into several distinct categories, each with characteristic energy requirements and structural implications:
σ → σ* transitions: These transitions involve the excitation of an electron from a bonding σ orbital to an antibonding σ* orbital. They typically require high energy (short wavelength) UV light, often below 200 nm, and are therefore not always observable in standard UV-Vis spectra (200-700 nm) [2] [11]. Molecules such as methane that contain only C-H bonds undergo primarily σ → σ* transitions [2].
n → σ* transitions: Saturated compounds containing atoms with lone pairs (non-bonding electrons) are capable of n → σ* transitions. These transitions usually need less energy than σ → σ* transitions and can be initiated by light in the 150-250 nm range [2] [11].
Ï€ → Ï€* transitions: Most absorption spectroscopy of organic compounds is based on transitions of Ï€ electrons to the Ï€* excited state, as these transitions typically fall in an experimentally convenient region of the spectrum (200-700 nm) [2]. These transitions require an unsaturated group in the molecule to provide the Ï€ electrons and generally give molar absorptivities between 1000 and 10,000 L molâ»Â¹ cmâ»Â¹ [2]. In conjugated systems, the energy gap for π→π* transitions decreases as conjugation increases, resulting in absorption at longer wavelengths [13].
n → Ï€* transitions: These transitions involve the excitation of a non-bonding electron (lone pair) to an antibonding Ï€* orbital. They are often observed in molecules with heteroatoms conjugated with Ï€ systems and require relatively low energy UV or visible light [13] [11]. Molar absorptivities for n→π* transitions are relatively low, typically ranging from 10 to 100 L molâ»Â¹ cmâ»Â¹ [2].
Charge-transfer transitions: Many inorganic species exhibit charge-transfer absorption, where absorption of radiation involves the transfer of an electron from a donor to an orbital associated with an acceptor [2] [11]. These transitions typically yield very large molar absorptivities (greater than 10,000 L molâ»Â¹ cmâ»Â¹) [2].
Selection Rules and Band Characteristics
The probability of electronic transitions is governed by selection rules derived from quantum mechanical principles. The spin selection rule states that transitions between states with different spin multiplicities are forbidden, making singlet-to-singlet and triplet-to-triplet transitions allowed, while singlet-to-triplet transitions are forbidden [11]. The Laporte selection rule (or parity selection rule) states that for centrosymmetric molecules, transitions involving a change in parity (g u) are allowed, while transitions with no change in parity (g g or u u) are forbidden [11].
These selection rules help explain why some transitions appear as weak absorption bands despite favorable energy matching. For instance, the weak absorption band of benzene at approximately 260 nm is attributed to a symmetry-forbidden π→π* transition that gains limited intensity through vibronic coupling [11].
Figure 1: UV-Vis Instrumentation and Electronic Transitions. This workflow illustrates the fundamental components of a UV-Vis spectrophotometer and the types of electronic transitions that occur during sample interaction.
Current Market Position and Technological Evolution
The UV-Vis spectroscopy market demonstrates robust growth and technological evolution, reflecting the technique's adaptability to modern analytical demands. By instrument type, benchtop spectrophotometers maintained a dominant 55.67% market share in 2024, while portable/hand-held devices are projected to expand at a notable 7.46% compound annual growth rate (CAGR) through 2030 [81]. This trend toward portability addresses growing needs for point-of-care testing, environmental field analysis, and process monitoring applications.
Technologically, instrumentation has evolved significantly from basic single-beam systems. While dual-beam optics continued to hold 41.45% revenue share in 2024, diode-array configurations logged a 7.76% CAGR and are positioned to outpace other designs through 2030 [81]. These configurations enable simultaneous capture of entire spectra in milliseconds, supporting advanced applications such as peak-purity checks and forced-degradation profiling [81].
Table 1: UV-Vis Spectroscopy Market Analysis by Segment (2024)
| Segment | Market Share | Growth Projection | Key Drivers |
|---|---|---|---|
| Instrument Type | |||
| Benchtop | 55.67% | Stable | Regulatory compliance, method transfer [81] |
| Portable/Hand-held | 18.32% | 7.46% CAGR | Point-of-care testing, field applications [81] |
| Technology | |||
| Dual-beam | 41.45% | Stable | Baseline stability for long analytical runs [81] |
| Diode-array | 28.91% | 7.76% CAGR | Rapid full-spectrum capture, peak purity [81] |
| Application | |||
| Drug Quality Control | 46.43% | Stable | Regulatory mandates for batch release [81] |
| Bioprocess Monitoring | 15.21% | 8.56% CAGR | Biologics pipeline growth, continuous processing [81] |
| End User | |||
| Pharmaceutical & Biotech | 48.54% | Stable | Quality by Design, biologics focus [81] |
| Academic & Research | 12.35% | 8.23% CAGR | Research infrastructure investment [81] |
Application analysis reveals that drug quality control and release testing commanded 46.43% of the UV-Vis spectroscopy market size in 2024, while bioprocess monitoring is forecast to grow at an impressive 8.56% CAGR to 2030 [81]. This shift reflects the biopharmaceutical sector's transition toward continuous processing and real-time quality monitoring [81].
Geographically, North America led with 42.34% of market share in 2024, yet Asia-Pacific is expected to post the fastest regional CAGR of 6.45% through 2030, driven by increasing industrialization, government investments in healthcare infrastructure, and growing analytical capabilities [81] [82].
Strengths of UV-Vis Spectroscopy
Operational Advantages
UV-Vis spectroscopy offers significant practical benefits that contribute to its enduring popularity in analytical laboratories:
User-friendly operation: Modern UV-Vis spectrophotometers feature intuitive interfaces that streamline operation from sample preparation to data analysis, making the technique accessible even to those with minimal training and reducing operational costs [83].
Cost-effectiveness: UV-Vis instrumentation offers a compelling advantage with respect to both initial investment and operational expenses compared to many other analytical techniques, with low maintenance requirements and durability contributing to long-term cost-effectiveness [83].
Rapid analysis: Spectrophotometers typically provide results in a matter of seconds, enabling high-throughput analysis essential for quality control environments and kinetic studies [83].
Non-destructive nature: UV-Vis is generally a non-destructive technique that allows the sample to be recovered and used for further analyses, a particularly valuable characteristic when working with precious or limited-quantity samples [80].
Analytical Performance
The analytical capabilities of UV-Vis spectroscopy make it particularly valuable for both research and routine applications:
Excellent quantitative capabilities: UV-Vis excels in precise and accurate quantitative measurements, offering high sensitivity and resolution for detecting minute changes in absorbance across various concentrations, from trace levels to saturation points [83]. The technique follows the Beer-Lambert law, which establishes a direct proportional relationship between absorbance and concentration, enabling precise quantitation [8] [80].
Broad application versatility: UV-Vis spectroscopy finds utility across diverse scientific disciplines including pharmaceutical analysis, biological research, environmental monitoring, food and beverage quality control, and materials science [8] [80]. This versatility stems from the fundamental nature of electronic transitions that are common to a wide range of chemical species.
High sensitivity for conjugated systems: Molecules with extended conjugated π systems exhibit strong, characteristic absorbance in the UV-Vis range, making the technique particularly sensitive for detecting and quantifying pharmaceuticals, dyes, natural products, and other conjugated compounds [13].
Limitations and Constraints
Despite its considerable strengths, UV-Vis spectroscopy exhibits several inherent limitations that analysts must consider when selecting appropriate analytical methodologies:
Limited structural information: UV-Vis spectra typically contain only a few broad absorption bands, making it difficult to derive detailed structural information or to distinguish between compounds with similar chromophores [2] [84]. This limitation contrasts with techniques like infrared spectroscopy or nuclear magnetic resonance, which provide more detailed molecular fingerprints.
Spectral overlap in mixtures: In complex matrices containing multiple absorbing species, significant spectral overlap can occur, complicating both qualitative identification and quantitative analysis without employing separation techniques or advanced chemometric approaches [84].
Solvent and environmental restrictions: The choice of solvent is critical in UV-Vis spectroscopy, as many common solvents absorb in the UV range, restricting available measurement windows [8] [2]. Additionally, air absorbs strongly below 200 nm, requiring specialized instrumentation for far-UV measurements [8].
Adherence to Beer-Lambert law limitations: The linear relationship between absorbance and concentration holds only within certain concentration ranges, with deviations occurring at high concentrations due to molecular interactions or instrumental factors such as stray light [8].
Susceptibility to interference: The technique can be affected by various interferents including turbidity (light scattering), gas bubbles, or temperature fluctuations that may impact absorbance measurements and introduce errors [8].
Table 2: Comparative Analysis of UV-Vis Spectroscopy with Alternative Techniques
| Parameter | UV-Vis Spectroscopy | HPLC | Fluorescence | Mass Spectrometry |
|---|---|---|---|---|
| Cost | Low to moderate | High | Moderate | Very high |
| Speed | Very fast (seconds) | Slow (minutes-hours) | Fast | Moderate |
| Sensitivity | Good (ppm-ppb) | Excellent (ppb-ppt) | Excellent (single molecule) | Outstanding (single molecule) |
| Structural Information | Limited | Moderate with standards | Moderate | Extensive |
| Quantitative Capability | Excellent | Excellent | Excellent | Good |
| Ease of Use | Very easy | Requires training | Moderate | Extensive training needed |
| Sample Throughput | High | Low to moderate | High | Moderate |
Methodological Framework and Experimental Protocols
Instrumentation and Components
Modern UV-Vis spectrophotometers consist of several key components, each serving a specific function in the measurement process:
Light sources: Instruments typically employ multiple light sources to cover the full UV-Vis range, commonly combining a deuterium lamp for UV light (190-400 nm) and a tungsten or halogen lamp for visible light (400-800 nm) [8] [80]. More expensive xenon lamps offer high intensity across both ranges but may suffer from stability issues [8].
Wavelength selection: Monochromators containing diffraction gratings are most commonly used for wavelength selection, separating light into narrow bandwidths with typical groove frequencies of 1200-2000 grooves per mm to balance resolution and wavelength range [8]. Interference filters and absorption filters may also be employed for specific applications [8].
Sample containers: Cuvettes with standard path lengths of 1 cm are most common, though path lengths from 0.01 mm to 100 mm are available for specialized applications [8] [84]. Quartz or fused silica cuvettes are required for UV measurements below 350 nm, as glass and plastic absorb significantly in this region [8].
Detection systems: Modern instruments employ various detectors including photomultiplier tubes (PMT), photodiodes, and charge-coupled devices (CCD) [8]. PMTs offer high sensitivity for low-light applications, while diode arrays and CCDs enable simultaneous capture of entire spectra [8].
Standard Experimental Protocol
A systematic approach ensures accurate and reproducible UV-Vis measurements:
Instrument calibration: Prior to analysis, perform wavelength and photometric accuracy verification using certified reference materials such as holmium oxide or didymium filters [81].
Sample preparation:
- Prepare samples in appropriate solvents that do not absorb significantly at wavelengths of interest
- Ensure analyte concentration falls within the instrument's linear dynamic range (typically absorbance values of 0.1-1.0 AU)
- For concentrated samples, employ path length reduction or dilution to maintain Beer-Lambert law compliance [8]
Reference measurement: Measure a blank containing only the solvent or matrix to establish baseline absorbance [8].
Data acquisition:
- Select appropriate spectral bandwidth based on analysis requirements
- Set scan speed to balance resolution and measurement time
- Acquire spectra over relevant wavelength range
Data analysis:
Figure 2: UV-Vis Experimental Workflow. This diagram outlines the standard methodology for UV-Vis analysis, highlighting critical considerations at each stage to ensure accurate results.
Advanced Methodologies: Chemometric Approaches
Modern UV-Vis analysis increasingly incorporates chemometric techniques to overcome traditional limitations, particularly for complex mixtures [84]. These mathematical and statistical methods extract meaningful information from chemical data through:
Spectral preprocessing: Techniques including smoothing, derivatives, multiplicative scatter correction, and standard normal variate transformation enhance spectral quality and reduce non-chemical variances [84].
Multivariate calibration: Methods such as Principal Component Regression (PCR) and Partial Least Squares (PLS) enable quantitative analysis of individual components in mixtures without physical separation by correlating spectral data with concentration information [84].
Pattern recognition: Both supervised (PLS-DA, SIMCA) and unsupervised (PCA, clustering) techniques facilitate classification and identification of samples based on their spectral fingerprints [84].
The integration of chemometrics has revitalized UV-Vis applications in challenging matrices, transforming the technique from a simple data provider to a comprehensive chemical information source capable of addressing complex analytical problems [84].
Essential Research Reagent Solutions
Table 3: Key Reagents and Materials for UV-Vis Spectroscopy
| Reagent/Material | Function/Purpose | Technical Specifications | Application Notes |
|---|---|---|---|
| Quartz Cuvettes | Sample containment for UV range | High-purity quartz, path lengths 0.01-100 mm | Required for <350 nm; transparent to ~200 nm [8] |
| Solvents (HPLC Grade) | Sample dissolution & dilution | UV-transparent, low impurity | Acetonitrile, water, hexane common; check UV cutoff [8] |
| Certified Reference Materials | Wavelength & photometric accuracy | Holmium oxide, didymium filters | Instrument validation per pharmacopeial standards [81] |
| Buffer Systems | pH control for biological samples | Phosphate, Tris, carbonate buffers | Ensure buffer transparency at analysis wavelength [8] |
| Standard Analytical Kits | Quantitative calibration | DNA/RNA, protein, cell density kits | Pre-validated methods for specific applications [80] |
UV-Vis spectroscopy maintains a vital position in the modern analytical toolkit, successfully balancing traditional reliability with contemporary innovations. The technique's future trajectory appears shaped by several key developments:
Miniaturization and portability: The growing demand for field-based analysis and point-of-care testing drives development of compact, handheld UV-Vis devices with performance characteristics approaching those of benchtop instruments [85] [81]. These portable systems increasingly incorporate wireless connectivity and cloud-based data management for enhanced operational flexibility [81].
Advanced detection systems: Continued improvement in detector technology, including enhanced photodiode arrays and CCD sensors, provides greater sensitivity, faster acquisition times, and improved signal-to-noise ratios [8] [84].
Integration with complementary techniques: UV-Vis spectroscopy increasingly functions as part of hyphenated systems, particularly as a detector for liquid chromatography, enabling comprehensive analysis that combines separation power with spectral characterization [83].
Intelligent data analysis: Artificial intelligence and machine learning algorithms are being integrated into UV-Vis data analysis workflows, enabling more efficient data interpretation, pattern recognition, and predictive modeling [84].
In conclusion, UV-Vis spectroscopy remains an indispensable analytical technique that has evolved to meet modern scientific demands. Its unique combination of operational simplicity, cost-effectiveness, and quantitative reliability ensures its continued relevance alongside more sophisticated analytical methods. While the technique exhibits inherent limitations in structural elucidation and specificity for complex mixtures, ongoing technological advancements—particularly in chemometrics, miniaturization, and detection systems—continue to expand its application boundaries. For researchers and analytical professionals, UV-Vis spectroscopy represents a foundational technique that provides the first insight into molecular electronic structure while offering the practical utility necessary for routine analysis across pharmaceutical, biological, environmental, and materials science disciplines.
The accurate quantification of chemical compounds and assessment of their purity are foundational to research and development in pharmaceuticals, environmental science, and materials science. Among the most prevalent techniques for these analyses are Ultraviolet-Visible (UV-Vis) Spectrophotometry and High-Performance Liquid Chromatography (HPLC). The fundamental theory underlying UV-Vis detection is based on the excitation of valence electrons in a molecule by ultraviolet or visible light, leading to electronic transitions from the ground state to an excited state [2] [13]. While both methods can leverage these electronic transitions for detection, they differ profoundly in their operational principles, capabilities, and applications. This guide provides an in-depth technical comparison of these two techniques, focusing on their use in quantification and purity assessment, supported by experimental data and current best practices.
Theoretical Foundations: Electronic Transitions in UV-Vis Spectroscopy
UV-Vis spectroscopy measures the absorption of light in the ultraviolet (200-400 nm) and visible (400-700 nm) regions of the electromagnetic spectrum. This absorption occurs when the energy of the incoming photons matches the energy required to promote an electron from its ground state to a higher energy state [13].
The primary electronic transitions involved are:
- Ï€ → Ï€* transitions: These occur in molecules with conjugated Ï€ systems. The energy gap for this transition decreases as conjugation increases, resulting in absorption at longer wavelengths. These transitions are typically strong, with molar absorptivities (ε) between 1,000 and 10,000 L·molâ»Â¹Â·cmâ»Â¹ [2].
- n → Ï€* transitions: These involve the excitation of a non-bonding (lone pair) electron into a Ï€* antibonding orbital. They are weaker than Ï€ → Ï€* transitions (ε = 10 to 100 L·molâ»Â¹Â·cmâ»Â¹) and often occur at longer wavelengths. The solvent polarity can cause a blue shift (shift to shorter wavelengths) for n → Ï€* transitions due to increased solvation of the lone pair electrons [2] [13].
The relationship between the absorbance of a solution and the concentration of the analyte is quantitatively described by the Beer-Lambert Law: ( A = \epsilon b c ), where ( A ) is the measured absorbance, ( \epsilon ) is the molar absorptivity, ( b ) is the path length, and ( c ) is the concentration [2]. This principle forms the basis for quantitative analysis using UV-Vis.
Diagram: Electronic Transitions in UV-Vis Spectroscopy
High-Performance Liquid Chromatography (HPLC): Principles of Separation
HPLC is a separation technique that distributes components of a chemical mixture between a stationary phase and a mobile phase [86]. The mobile phase is a liquid pumped at high pressure through a column packed with a stationary phase. Separation occurs because different analytes have varying degrees of interaction with the stationary phase. Compounds with stronger interactions with the stationary phase are retained longer in the column, resulting in a longer retention time, while those with a greater affinity for the mobile phase elute faster [87] [86].
The core separation modes in HPLC include:
- Reversed-Phase Chromatography (RP): The most common mode, which separates components based on hydrophobicity [87].
- Normal-Phase Chromatography (NP): Separates components based on hydrophilicity or polarity [87].
- Ion Exchange Chromatography (IEX): Utilizes electrostatic interactions for separation [87].
- Size Exclusion Chromatography (SEC): Separates molecules based on their size [87].
Detection in HPLC is often achieved using a UV-Vis detector, which applies the same principles of electronic transitions described above. However, the key advantage of HPLC is that it separates the mixture into individual components before detection, allowing for the specific quantification and purity assessment of each analyte.
Diagram: Basic Workflow of an HPLC System
Comparative Analysis: UV-Vis vs. HPLC
The choice between UV-Vis and HPLC depends heavily on the analytical goals, sample complexity, and required data rigor. The table below summarizes their core characteristics.
Table 1: Core Characteristics of UV-Vis and HPLC
| Feature | UV-Vis Spectrophotometry | High-Performance Liquid Chromatography (HPLC) |
|---|---|---|
| Basic Principle | Measures electronic transitions of molecules in solution [2]. | Separates compounds via differential partitioning between mobile and stationary phases, then detects them (often via UV) [87] [86]. |
| Primary Application | Direct quantification of a single, pure analyte or the total chromophore content in a simple mixture. | Separation, identification, and quantification of individual components in a complex mixture. |
| Key Strength | Rapid, simple, cost-effective, and high-throughput for suitable samples. | High selectivity, resolution, and ability to handle complex samples. |
| Purity Assessment | Limited and indirect; relies on spectral shape or absorbance ratios, assumes sample purity [88]. | Direct and robust; uses chromatographic resolution and peak purity algorithms (e.g., PDA) to detect co-eluting impurities [88]. |
| Sensitivity | Good (depends on ε). | Excellent, especially with advanced detectors (e.g., MS). |
| Sample Throughput | Very high. | Moderate to high (faster with UHPLC). |
| Cost & Operational Complexity | Low cost, simple operation. | High cost, requires skilled personnel and method development [89]. |
Quantitative Performance and Purity Assessment
A direct comparative study on Levofloxacin analysis highlights the practical differences in quantitative performance. The study established standard curves for both techniques, showing both could achieve excellent linearity (R² > 0.999) [90]. However, a critical difference emerged in the accuracy, as measured by recovery rates from a complex matrix (simulated body fluid).
Table 2: Quantitative Performance Data for Levofloxacin Analysis [90]
| Parameter | HPLC Method | UV-Vis Method |
|---|---|---|
| Regression Equation | y = 0.033x + 0.010 | y = 0.065x + 0.017 |
| Linearity (R²) | 0.9991 | 0.9999 |
| Recovery (Low Conc.) | 96.37 ± 0.50% | 96.00 ± 2.00% |
| Recovery (Medium Conc.) | 110.96 ± 0.23% | 99.50 ± 0.00% |
| Recovery (High Conc.) | 104.79 ± 0.06% | 98.67 ± 0.06% |
The variable and sometimes excessive recovery rates observed with HPLC in this specific study can be context-dependent. However, the key finding was that UV-Vis was deemed not accurate for measuring drugs loaded on biodegradable composite scaffolds due to interference from other scaffold components that also absorbed UV light. The study concluded that HPLC is the preferred method for evaluating the sustained-release characteristics of drugs from complex matrices because of its superior selectivity [90].
For purity assessment, HPLC with a Photodiode Array (PDA) detector is the industry standard. Peak Purity Assessment (PPA) software compares UV spectra across a chromatographic peak. A pure peak will have a consistent spectrum, while a co-eluting impurity will cause spectral variations [88]. While powerful, PDA-based PPA has limitations, including potential for false negatives if an impurity has a nearly identical UV spectrum to the main compound or is present at a very low concentration [88]. For ultimate confidence, mass spectrometry (MS) is used as a detector, as it can differentiate components based on molecular mass [91] [88].
Experimental Protocols
Detailed Methodology: Comparative Analysis of Levofloxacin
The following protocol is adapted from the study providing the data in Table 2 [90].
1. Instrumentation and Reagents:
- HPLC System: Shimadzu LC-2010AHT system with a UV detector and a Sepax BR-C18 column (250 × 4.6 mm, 5 µm).
- UV-Vis Spectrophotometer: Shimadzu UV-2600.
- Chemicals: Levofloxacin standard, Ciprofloxacin (internal standard), Methanol (HPLC-grade), Potassium dihydrogen phosphate (KHâ‚‚POâ‚„), Tetrabutylammonium hydrogen sulphate, Simulated Body Fluid (SBF).
2. Chromatographic Conditions (HPLC):
- Mobile Phase: A mixture of 0.01 mol/L KHâ‚‚POâ‚„, methanol, and 0.5 mol/L tetrabutylammonium hydrogen sulphate (75:25:4, v/v/v).
- Flow Rate: 1.0 mL/min.
- Detection Wavelength: 290 nm.
- Column Temperature: 40 °C.
- Injection Volume: 10 µL for assay.
3. Standard Solution Preparation:
- Precisely weigh 30.00 mg of Levofloxacin and dissolve in SBF in a 10 mL volumetric flask to make a 3 mg/mL stock solution.
- Serially dilute the stock solution with SBF to create at least 8 concentration levels across the working range (e.g., from 0.05 to 300 µg/mL).
4. Sample Preparation for HPLC:
- To 100 µL of the standard or sample solution, add 10 µL of the internal standard solution (Ciprofloxacin, 500 µg/mL in methanol).
- Vortex-mix the solution for 5 minutes.
- Add 800 µL of dichloromethane, vortex-mix for another 5 minutes.
- Centrifuge the mixture at ~7,155 × g for 5 minutes at 25°C.
- Transfer 750 µL of the supernatant, evaporate to dryness under a stream of nitrogen in a 50°C water bath.
- Reconstitute the dry residue with 100 µL of mobile phase and inject into the HPLC system.
5. Analysis by UV-Vis:
- Directly analyze the standard and sample solutions in SBF using a quartz cuvette in the UV-Vis spectrophotometer.
- Set the instrument to measure absorbance at the predetermined λ_max for Levofloxacin (e.g., 290 nm). Ensure the calibration curve is constructed using the same solvent (SBF).
6. Data Analysis:
- Construct calibration curves (Absorbance vs. Concentration for UV-Vis; Peak Area Ratio vs. Concentration for HPLC).
- Calculate the regression equation and correlation coefficient (R²).
- Determine the concentration of unknown samples using the regression equation.
- Calculate recovery rates for accuracy validation.
Key Research Reagent Solutions
Table 3: Essential Materials for HPLC and UV-Vis Analysis
| Item | Function/Description |
|---|---|
| C18 Chromatographic Column | The most common reversed-phase stationary phase for separating mid- to non-polar analytes [90] [86]. |
| HPLC-Grade Solvents | High-purity solvents (e.g., methanol, acetonitrile, water) used for the mobile phase to minimize baseline noise and prevent column damage. |
| Buffer Salts | Salts (e.g., KHâ‚‚POâ‚„, ammonium acetate) are used to control the pH and ionic strength of the mobile phase, critical for separating ionizable compounds [90]. |
| Standard/Internal Standard | A high-purity reference compound of the target analyte for quantification and a similar compound to correct for procedural variability [90]. |
| Simulated Body Fluid (SBF) | A buffer solution that mimics the ionic composition of human blood plasma, used for in-vitro release studies [90]. |
Recent Advancements and Future Trends
The fields of UV-Vis and HPLC are continuously evolving, with recent trends focusing on efficiency, sensitivity, and connectivity.
- UV-Vis Evolution: Modern UV-Vis instruments in 2025 emphasize user-friendly interfaces with touchscreens and guided workflows, smaller footprints to save lab space, and faster scanning speeds for higher throughput. Improved optical stability and thermal regulation ensure consistent, reliable results with less calibration [92].
- HPLC/UHPLC Innovation: New systems introduced in 2024-2025, such as Shimadzu's i-Series and Thermo Fisher's Vanquish Neo, highlight compact designs, higher pressure capabilities (e.g., 70-1300 bar), and eco-friendly features with reduced energy and solvent consumption [93]. There is a strong push towards intelligent automation, integration with LC-MS/MS for definitive identification, and the use of green chromatography principles to reduce environmental impact [93] [89].
- Advanced Detection Techniques: For complex purity challenges, 2D-LC and Mass Spectrometry-facilitated peak purity are becoming more accessible. MS detection provides unequivocal identification of co-eluting impurities based on mass, overcoming the limitations of UV spectral comparison [88].
UV-Vis spectrophotometry and HPLC are complementary but not interchangeable techniques. UV-Vis is a powerful, straightforward tool for the quantitative analysis of pure chromophores or the study of electronic transitions in simple systems. However, its utility is severely limited in complex matrices where selectivity is required. HPLC, with its powerful separation capability coupled with UV (PDA) or MS detection, is the unequivocal method of choice for reliable quantification in complex samples and for rigorous purity assessment. The decision between them must be guided by the nature of the sample, the required information, and the necessary level of analytical confidence.
Cross-Validation with 1H NMR for Structural Confirmation and Quality Control
The fundamental theory of electronic transitions in UV-Vis spectroscopy establishes that molecular electrons absorb specific wavelengths of light when promoted from ground state to excited state orbitals. The energy required for these transitions, particularly the HOMO-LUMO (Highest Occupied Molecular Orbital to Lowest Unoccupied Molecular Orbital) gap, determines the absorption characteristics of chromophores [4] [6]. While UV-Vis spectroscopy directly probes these electronic transitions, Nuclear Magnetic Resonance (NMR) spectroscopy provides complementary information about the local chemical environment surrounding atomic nuclei. Cross-validation using 1H NMR leverages this detailed structural information to confirm molecular identity and purity, connecting electronic structure properties from UV-Vis with atomic-level structural verification for comprehensive quality control in pharmaceutical development and other scientific fields.
The integration of these analytical techniques provides a powerful framework for structural elucidation. UV-Vis identifies chromophoric systems through their electronic transition patterns, while 1H NMR validates the specific molecular framework hosting these chromophores. This dual approach is particularly valuable for quality control, where both the electronic properties (relevant for photodegradation studies) and structural integrity must be verified simultaneously [4].
Theoretical Foundations: From Electronic Transitions to Nuclear Spin
Electronic Transitions in UV-Vis Spectroscopy
Ultraviolet-Visible spectroscopy operates on the principle that molecules contain electrons in various molecular orbitals at distinct energy levels. When exposed to UV or visible light, these electrons can absorb photons with energy matching exactly the difference between two orbital energy levels (ΔE), causing electronic transitions from ground state to excited state [4]. The energy of these transitions follows the equation: [ E = hν = \frac{hc}{λ} ] where (E) is energy, (h) is Planck's constant, (ν) is frequency, (c) is the speed of light, and (λ) is wavelength [4].
Key electronic transitions include:
- σ→σ* transitions: Require high energy, typically occurring in the far-UV region (<150 nm) for single bonds [4]
- π→π* transitions: Found in molecules with double bonds and conjugated systems, occurring at longer wavelengths (165-250 nm) [4]
- n→π* transitions: Involve promotion of non-bonding electrons to π* orbitals, typically observed at longer wavelengths (250-300 nm) [13]
The extent of conjugation significantly affects these transitions. As conjugation increases, the HOMO-LUMO energy gap decreases, resulting in bathochromic shifts (absorption at longer wavelengths) [4] [6]. This principle explains why highly conjugated compounds like β-carotene absorb visible light and appear colored [4].
Principles of 1H NMR Spectroscopy
While UV-Vis spectroscopy examines electronic transitions, 1H NMR spectroscopy probes the magnetic properties of atomic nuclei. When placed in a strong magnetic field, 1H nuclei (protons) can exist in different spin states. Absorption of radiofrequency radiation promotes transitions between these nuclear spin states, providing detailed information about the chemical environment of each proton [94] [95].
The key parameters in 1H NMR include:
- Chemical shift (δ): Reflects the electronic environment of a proton, measured in parts per million (ppm)
- Integration: Proportional to the number of equivalent protons
- Spin-spin coupling: Provides information about adjacent protons through splitting patterns
For quality control applications, 1H NMR serves as a powerful fingerprinting technique because it can simultaneously identify and quantify multiple compounds in complex mixtures without requiring extensive separation [95].
NMR Validation Methodologies for Structural Confirmation
The ANSURR Method for Protein Structure Validation
The Accuracy of NMR Structures using Random Coil Index and Rigidity (ANSURR) method represents an advanced approach for validating protein structures determined by NMR. This technique compares two independent measures of protein flexibility [94]:
- Experimental flexibility derived from backbone chemical shifts using the Random Coil Index (RCI)
- Predicted flexibility calculated from the protein structure using mathematical rigidity theory with the FIRST software
The ANSURR method generates two primary scores:
- Correlation score: Assesses whether rigid and flexible regions align properly, primarily validating secondary structure elements
- RMSD score: Measures overall rigidity matching, sensitive to hydrogen bonding networks and sidechain interactions [94]
This validation approach addresses a critical limitation in NMR structure determination: the lack of equivalent metrics to crystallographic R-factors for assessing accuracy against experimental data [94]. Traditional NMR validation metrics like restraint violations and ensemble RMSD provide information about precision and restraint satisfaction but offer limited insight into actual structural accuracy [94].
Quantitative NMR for Compound Validation
Quantitative 1H NMR (qNMR) provides a robust methodology for validating chemical structures and determining purity. The technique relies on the direct proportionality between signal intensity and the number of nuclei generating the signal [95]. For structural confirmation, qNMR can:
- Verify molecular identity through characteristic chemical shifts
- Determine purity using internal standards
- Quantify components in mixtures without reference materials
- Detect and identify impurities at levels as low as 0.1%
The PULCON (pulse length-based concentration determination) method enables accurate quantification by correlating signal intensities with a reference of known concentration [95]. This approach has been validated for various applications including the analysis of coffee components, where it simultaneously quantifies caffeine, 16-O-methylcafestol (OMC), kahweol, furfuryl alcohol, and 5-hydroxymethylfurfural (HMF) in a single experiment [95].
NMR in Quality Control Applications
Food and Agricultural Products
1H NMR spectroscopy has demonstrated exceptional utility in quality control for food products, particularly for authentication and adulteration detection. In coffee analysis, NMR simultaneously quantifies multiple markers that differentiate coffee species and assess quality [95]:
Table 1: NMR Quality Markers in Coffee Analysis
| Compound | Role in Quality Control | Typical Concentration Range | Significance |
|---|---|---|---|
| 16-O-methylcafestol (OMC) | Authenticity marker | <50 mg/kg for arabica | Specific to Coffea canephora (robusta) |
| Caffeine | Quality marker | <1000 mg/kg for decaf | Compliance with regulations |
| Kahweol | Authenticity marker | <300 mg/kg for robusta | Higher in Coffea arabica |
| Furfuryl alcohol | Process indicator | Variable | Indicator of roasting degree |
| 5-HMF | Process indicator | Variable | Thermal processing marker |
This multi-parameter approach enables comprehensive quality assessment. For instance, detecting OMC in products labeled as 100% arabica coffee indicates adulteration with the cheaper robusta species [95]. Similarly, elevated levels of furfuryl alcohol and HMF may indicate over-roasting or improper storage conditions [95].
Bio-wax and Industrial Products
For bio-waxes derived from vegetable oils, 1H NMR coupled with chemometric analysis predicts key chemical properties including acid value (AV), saponification value (SV), and iodine value (IV) [96]. This approach eliminates the need for traditional wet chemistry methods that use toxic reagents, require long analysis times, and generate significant waste [96].
The methodology involves:
- Acquiring 1H NMR spectra of bio-wax samples
- Identifying characteristic signals for different components (paraffins, free fatty acids, fatty alcohols, esters, glycerides)
- Applying multivariate calibration models (PCR, PLSR) to correlate spectral data with reference values
- Validating models using cross-validation and external test sets
This NMR-based approach successfully predicts chemical properties with precision comparable to standard methods while offering advantages in speed, cost, and environmental impact [96].
Experimental Protocols and Methodologies
Sample Preparation for Quantitative NMR
Proper sample preparation is critical for reliable quantitative NMR results. The following protocol is adapted from validated methods for coffee analysis [95]:
- Weighing: Accurately weigh 200 mg of ground sample material
- Extraction: Add 1.5 mL of deuterated chloroform with 1% tetramethylsilane (TMS) as internal standard
- Agitation: Shake at 350 rpm for 10-20 minutes using a mechanical shaker
- Filtration: Pass solution through 0.45 μm membrane filter to remove particulate matter
- Transfer: Pipette 600 μL of filtrate into a standard 5 mm NMR tube
For complex matrices, optimization of extraction time and solvent composition may be necessary. A factorial experimental design can systematically evaluate the influence of different parameters (extraction time, solvent composition, temperature) on extraction efficiency [95].
NMR Instrumentation and Acquisition Parameters
Standardized acquisition parameters ensure reproducibility and quantitative reliability [95]:
Table 2: Quantitative 1H NMR Acquisition Parameters
| Parameter | Setting | Purpose |
|---|---|---|
| Field Strength | 400 MHz (9.4 T) | Balance between sensitivity and resolution |
| Pulse Sequence | zg30 (single pulse) | Minimize relaxation effects |
| Relaxation Delay (D1) | 30 s | Ensure complete T1 recovery |
| Number of Scans (NS) | 64 | Improve signal-to-noise ratio |
| Acquisition Time | 7.97 s | Adequate digitization of FID |
| Spectral Width | 20.55 ppm | Cover entire chemical shift range |
| Temperature | 300.0 K | Maintain stability |
| Window Function | 0.30 Hz line broadening | Optimize sensitivity/resolution balance |
The prolonged relaxation delay (5× typical T1 values) ensures complete longitudinal relaxation between scans, which is essential for quantitative accuracy [95]. The PULCON method correlates signal intensities with an external reference for concentration determination [95].
Data Processing and Multivariate Analysis
For complex mixtures, multivariate statistical methods enhance the information extracted from NMR spectra:
- Spectral preprocessing: Phase correction, baseline correction, and chemical shift referencing to TMS (0 ppm)
- Data reduction: Segment spectra into bins (e.g., 0.04 ppm width) or select characteristic peaks
- Model development: Apply Principal Component Regression (PCR) or Partial Least Squares Regression (PLSR) to correlate spectral features with reference values
- Validation: Use cross-validation and external test sets to evaluate prediction accuracy [96]
This approach has successfully predicted acid, saponification, and iodine values of bio-waxes with R² values exceeding 0.9 for calibration and validation sets [96].
Diagram: NMR Cross-Validation Workflow
NMR Cross-Validation Workflow for Quality Control - This diagram illustrates the integrated approach combining 1H NMR and UV-Vis spectroscopy for structural confirmation and quality assessment, highlighting the complementary nature of these techniques.
Essential Research Reagents and Materials
Table 3: Essential Research Reagents for NMR Cross-Validation
| Reagent/Material | Specifications | Function in Analysis |
|---|---|---|
| Deuterated Solvents | CDCl₃, DMSO-d₆, D₂O (≥99.8% D) | NMR solvent providing lock signal |
| Internal Reference | Tetramethylsilane (TMS) | Chemical shift reference (0 ppm) |
| Quantitative Standards | Certified reference materials | Method calibration and validation |
| NMR Tubes | 5 mm, DEU-Quant precision | Sample containment for NMR analysis |
| Sample Filters | 0.45 μm membrane | Particulate removal for clear solutions |
| Quality Control Materials | Certified quality control samples | System suitability testing |
Cross-validation with 1H NMR represents a powerful approach for structural confirmation and quality control that complements electronic transition data from UV-Vis spectroscopy. By integrating information about local chemical environments (NMR) with electronic structure characteristics (UV-Vis), this dual methodology provides comprehensive molecular verification essential for pharmaceutical development, food authentication, and industrial product quality assurance. The quantitative nature of 1H NMR, coupled with advanced validation methods like ANSURR for proteins and multivariate calibration for complex mixtures, enables precise structural confirmation and purity assessment that meets rigorous quality control standards across research and industrial applications.
Ultraviolet-Visible (UV-Vis) spectroscopy is a powerful analytical technique that measures the absorption of light in the ultraviolet and visible regions of the electromagnetic spectrum. The fundamental principle underlying this technique involves electronic transitions, where molecules absorb specific wavelengths of light that provide precisely the energy needed to promote electrons from ground states to excited states [2]. When analyzing polymers and biomaterials, these electronic transitions provide critical insights into molecular structure, conjugation extent, and degradation pathways that directly impact material performance and stability.
The absorption of UV or visible radiation corresponds to the excitation of outer electrons, with several transition types being particularly relevant for polymeric materials. These include transitions involving π, σ, and n electrons and charge-transfer electrons [2]. In conjugated polymers, the most significant transitions typically occur between π and π* orbitals (π→π* transitions), where the energy gap correlates directly with the extent of conjugation within the molecular backbone [6]. The measurable relationship between absorbed light and sample properties is quantitatively described by the Beer-Lambert Law (A = εbc), which establishes that absorbance (A) is proportional to the concentration (c) of the absorbing species and the path length (b) of the measurement cell [8].
This technical guide explores the application of UV-Vis spectroscopy for characterizing conjugation and degradation in polymers and biomaterials, with emphasis on experimental methodologies, data interpretation, and practical applications within research and development contexts.
Theoretical Foundation: Electronic Transitions in Molecular Systems
Molecular Orbitals and Chromophores
At the heart of UV-Vis spectroscopy lie electronic transitions between molecular orbitals. These orbitals represent the probability distribution of electrons in a molecule, with bonding orbitals possessing lower energy than antibonding orbitals [6]. In the ground state, electrons typically occupy the lower-energy bonding orbitals, with the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) representing the frontier orbitals critical to light absorption phenomena [6].
Chromophores, the light-absorbing components of molecules, contain valence electrons of relatively low excitation energy. Common chromophores in polymer systems include C=C double bonds, carbonyl groups (C=O), and aromatic rings [2] [6]. When incorporated into conjugated systems where single and double bonds alternate, electron delocalization occurs across multiple bonds, significantly reducing the energy gap between HOMO and LUMO orbitals. This reduced energy gap manifests as absorption at longer wavelengths, a phenomenon known as a bathochromic shift [6].
Types of Electronic Transitions
Several types of electronic transitions are particularly relevant to polymer and biomaterial analysis:
- π→π* transitions: These occur in molecules with unsaturated centers such as C=C, C≡C, and aromatic rings. They typically yield high molar absorptivities (ε = 1,000-10,000 L·molâ»Â¹Â·cmâ»Â¹) and are often observed in conjugated polymers [2] [6].
- n→π* transitions: These involve the excitation of non-bonding electrons (lone pairs) to Ï€* antibonding orbitals, commonly found in carbonyl-containing polymers. They display lower molar absorptivities (ε = 10-100 L·molâ»Â¹Â·cmâ»Â¹) and often undergo blue shifts with increasing solvent polarity [2].
- Charge-transfer transitions: These occur when electron donation from a donor to an acceptor component takes place within a complex, resulting in very high molar absorptivities (ε > 10,000 L·molâ»Â¹Â·cmâ»Â¹) [2].
The energy required for these transitions follows the order: σ→σ* > n→σ* > π→π* > n→π*, determining the specific wavelength regions where different molecular features absorb light [2].
Spectral Shifts and Their Interpretation
Changes in molecular structure and environment manifest as shifts in UV-Vis absorption spectra:
- Bathochromic shift (red shift): Movement of absorption to longer wavelengths, often resulting from increased conjugation, electron-donating substituents, or solvent effects that stabilize excited states [6].
- Hypsochromic shift (blue shift): Movement of absorption to shorter wavelengths, caused by factors that increase the HOMO-LUMO gap, such as decreased conjugation, electron-withdrawing groups, or solvent effects that destabilize excited states [6].
- Hyperchromic effect: Increase in absorption intensity, often indicating enhanced transition probability or molecular ordering.
- Hypochromic effect: Decrease in absorption intensity, potentially signaling molecular aggregation or conformational changes.
These spectral perturbations provide critical insights into structural modifications during polymer degradation or changes in conjugation extent.
Diagram 1: Fundamental process of electronic transitions in UV-Vis spectroscopy, showing the pathway from photon absorption to spectral output and key influencing factors.
Characterizing Conjugation in Polymers
Conjugation Length and Spectral Properties
The extent of conjugation in polymeric systems directly influences their electronic structure and optical properties. As conjugation length increases, the HOMO-LUMO energy gap decreases, resulting in a bathochromic shift of the absorption maximum (λmax) [6]. This relationship enables researchers to use UV-Vis spectroscopy to quantitatively assess conjugation length in conducting polymers, semiconducting polymers, and other π-conjugated systems.
For example, in conjugated polymers like poly(3-hexylthiophene) (P3HT) and poly(3-hexylselenophene) (P3HS), UV-Vis spectroscopy effectively monitors aggregation dynamics through changes in the average photon energy (Eave) [97]. During J-aggregate formation, Eave decreases exponentially with time, providing a quantitative metric for tracking molecular organization and self-assembly processes [97].
Experimental Approach: Infiltration Kinetics in Nanoporous Scaffolds
Recent research demonstrates innovative applications of UV-Vis spectroscopy for characterizing polymer conformation and infiltration behavior. A 2025 study investigated the infiltration kinetics of poly(2-vinylpyridine) (P2VP) into nanoporous gold (NPG) scaffolds using UV-Vis spectroscopy [98]. The experimental protocol involved:
- Sample Preparation: NPG films with controlled pore radius (34 nm) were prepared as substrates for polymer infiltration.
- Thermal Processing: P2VP with varying molecular weights (85-940 kDa) was infiltrated into NPG at 140°C.
- Spectral Monitoring: UV-Vis spectra were collected at regular intervals during infiltration, tracking changes in plasmon absorption peaks characteristic of the Au ligaments.
- Data Analysis: The infiltration extent (IE) was correlated with spectral parameters including absorption peak position, peak height, width, and area [98].
This methodology revealed that the time to reach 80% infiltration (Ï„80%) scaled with molecular weight as Mw¹.³âµ, demonstrating the utility of UV-Vis spectroscopy for quantifying polymer diffusion in confined environments [98].
Table 1: UV-Vis Spectral Parameters for Monitoring Polymer Infiltration into Nanoporous Gold Scaffolds
| Spectral Parameter | Trend During Infiltration | Structural Interpretation | Measurement Precision |
|---|---|---|---|
| Absorption Peak Position | Shift to longer wavelengths | Increasing effective refractive index within pores | ±0.5 nm |
| Peak Height | Monotonic increase | Progressive filling of nanopores | ±2% |
| Peak Width | Monotonic increase | Enhanced light scattering from filled pores | ±1% |
| Peak Area | Monotonic increase | Cumulative infiltration extent | ±1.5% |
Quantitative Analysis of Aggregation Dynamics
The aggregation behavior of conjugated polymers significantly influences their optoelectronic properties. UV-Vis spectroscopy provides a powerful approach for quantifying these dynamics through analysis of spectral evolution. Research on P3HS and P3HT solutions has demonstrated that the changing rate of average photon energy (ΔEave) follows an exponential decay model during J-aggregate formation [97]. This parameter can predict both the completion time of aggregation and the progress toward completion at any measurement point [97].
Furthermore, the initial changing rate of Eave (ΔEâ°ave) reflects the overall aggregation trend and provides insights into solvent-solute compatibility, with higher values indicating more favorable conditions for aggregation [97]. The Hansen Solubility Parameters of solvents can even predict Eave values for conjugated polymer solutions without extensive experimental testing [97].
Monitoring Polymer Degradation
Photodegradation Mechanisms and Spectral Manifestations
Polymer degradation under UV exposure involves complex photochemical processes including chain scission, cross-linking, and formation of chromophoric groups such as carbonyls and hydroperoxides [99]. These chemical modifications alter the electronic structure of polymers, producing detectable changes in UV-Vis absorption spectra that serve as indicators of degradation extent.
For polyethylene (PE) and polypropylene (PP) automotive components, UV-induced degradation produces measurable changes in visible reflectance spectra across multiple wavelength bands [99]. Statistical analysis of reflectance data at 21 wavelengths grouped into seven visible bands (violet to red) enables quantitative comparison of degradation behavior under different environmental conditions [99].
Experimental Protocol: Assessing Cosmetic Degradation
A comprehensive approach for evaluating polymer degradation through UV-Vis spectroscopy involves:
- Sample Preparation: Prepare polymer specimens with consistent dimensions and surface characteristics. For automotive applications, this typically involves injection-molded plaques of PE and PP [99].
- Exposure Conditions: Subject samples to controlled degradation conditions including:
- No exposure (control)
- Outdoor exposure with glass protection
- Outdoor exposure without protection
- Accelerated UV chamber weathering (ASTM G154) [99]
- Spectral Measurement: Collect reflectance spectra at predetermined time intervals using a UV-Vis spectrophotometer equipped with integrating sphere attachment.
- Data Processing: Summarize reflectance data into defined visible bands (Violet to Red), with each band comprising three closely spaced wavelengths to serve as statistical replicates [99].
- Statistical Analysis: Employ repeated-measures ANOVA/MANOVA to evaluate effects of material, exposure condition, and time on optical properties, with initial reflectance values as covariates [99].
This protocol enables quantification of degradation efficiency η(t) through exponential modeling of spectral trajectories, providing a dimensionless parameter that summarizes degradation extent [99].
Table 2: UV-Vis Spectroscopy Applications in Polymer Degradation Studies
| Polymer System | Degradation Type | Key Spectral Changes | Analytical Approach |
|---|---|---|---|
| Polyethylene (PE) [99] | Photo-oxidation | Reflectance decay across visible bands | Multivariate statistical analysis |
| Polypropylene (PP) [99] | UV weathering | Band-dependent reflectance variability | Exponential modeling of spectral trajectories |
| Conjugated Polymers [100] | UV-Ozone treatment | Photobleaching, reduced semiconducting behavior | Absorption coefficient calculation |
| Polystyrene [101] | UVC sterilization | Minimal carbonyl formation | FT-IR correlation analysis |
| Wood-Plastic Composites [99] | Surface degradation | Reflectance shifts in visible range | Colorimetric analysis |
Case Study: Automotive Polymer Durability
Research on automotive PE and PP components demonstrates the practical application of UV-Vis spectroscopy for predicting service life. Studies employing a multi-statistical pragmatic framework have revealed significant effects of exposure condition, time, and their interaction on reflectance properties [99]. PE typically exhibits more gradual and coherent reflectance decay, while PP shows greater band-to-band variability, particularly under accelerated UV chamber exposure [99].
These spectral changes correlate with mechanical property degradation, as evidenced by decreased Shore D hardness in most exposed samples [99]. The combination of spectral and mechanical data validates UV-Vis spectroscopy as a non-destructive method for predicting long-term polymer performance in automotive applications.
Advanced Applications in Biomaterials and Nanoplastics
Surface Modification of Bioelectronic Polymers
UV-Vis spectroscopy plays a crucial role in optimizing biomaterial interfaces, particularly for organic semiconducting polymers used in bioelectronics. Research on pDPP3T films demonstrates how UV-ozone (UVO) treatment modifies surface properties to enhance cell adhesion while monitoring optical changes that might compromise functionality [100].
The experimental protocol involves:
- UVO Treatment: Expose polymer films to UVO for varying durations (0-30+ seconds).
- Surface Characterization: Assess polarity changes through contact angle measurements and XPS analysis.
- Optical Monitoring: Track photobleaching and reduction in semiconducting behavior through UV-Vis absorption spectra.
- Biological Validation: Evaluate Schwann cell growth and viability on treated surfaces [100].
This approach identifies an optimal treatment window (≥30 seconds) that enhances biointerfacing properties without significantly compromising electronic functionality [100].
Quantification of Environmental Nanoplastics
UV-Vis spectroscopy provides a rapid, accessible method for quantifying true-to-life nanoplastics in environmental research. A 2025 study established protocols for quantifying polystyrene-based nanoplastics generated from fragmented plastic items [102]:
- Nanoplastic Generation: Mechanically fragment polystyrene items under cryogenic conditions using an ultracentrifugal mill.
- Size Fractionation: Separate nanoplastics from microplastics through sequential centrifugation in MilliQ water.
- Spectroscopic Quantification: Analyze suspensions using microvolume UV-Vis spectrophotometry to minimize sample consumption.
- Method Validation: Compare results with established techniques including pyrolysis GC-MS, thermogravimetric analysis, and nanoparticle tracking analysis [102].
This methodology enables quantification of nanoplastic concentrations while conserving scarce samples for additional analyses, addressing a critical challenge in environmental nanotoxicology [102].
Diagram 2: Comprehensive experimental workflow for UV-Vis analysis of polymers, showing key steps from sample preparation through data interpretation.
Experimental Protocols and Methodologies
Standard Operating Procedure: Polymer Infiltration Kinetics
Based on recent research, the following detailed protocol characterizes polymer infiltration into nanoporous scaffolds:
Substrate Preparation:
- Fabricate nanoporous gold (NPG) scaffolds with controlled pore size (e.g., 34 nm radius) through dealloying of AgAu alloys.
- Characterize pore structure using SEM and surface area analysis.
Polymer Preparation:
- Select polymers with varying molecular weights (e.g., P2VP from 85-940 kDa).
- Purify polymers to remove residual monomers and additives.
Infiltration Process:
- Heat NPG-polymer assembly to specified temperature (140°C for P2VP).
- Maintain isothermal conditions throughout infiltration.
Spectral Monitoring:
- Collect UV-Vis spectra at regular intervals using a spectrophotometer with temperature control.
- Monitor plasmon absorption peaks between 400-800 nm.
- Record peak position, height, width, and area for each time point.
Data Analysis:
- Correlate spectral changes with infiltration extent using established models.
- Determine kinetic parameters (e.g., Ï„80%) for different molecular weights.
- Validate with complementary techniques (AFM, XPS) when possible [98].
Protocol: Degradation Monitoring for Automotive Polymers
For assessing UV degradation in automotive applications:
Sample Preparation:
- Obtain injection-molded plaques of PE and PP (typical dimensions: 50mm × 50mm × 3mm).
- Clean surfaces with isopropanol and allow to dry.
Experimental Design:
- Assign samples to exposure conditions (n≥4 per group):
- C1: Control (no exposure)
- C2: Outdoor with glass protection
- C3: Outdoor without protection
- C4: Accelerated UV chamber (ASTM G154)
- Include replicates for statistical power.
- Assign samples to exposure conditions (n≥4 per group):
Spectral Measurements:
- Measure initial (T0) reflectance spectra across 21 wavelengths in visible range.
- Group wavelengths into seven bands (Violet to Red) with three wavelengths per band.
- Repeat measurements at designated intervals (T1-T4).
- Use spectrophotometer with integrating sphere for consistent reflectance data.
Supplementary Analysis:
- Measure Shore D hardness pre- and post-exposure.
- Document visual changes through photography.
Statistical Analysis:
- Perform repeated-measures ANOVA/MANOVA with initial reflectance as covariate.
- Fit exponential models to spectral trajectories.
- Calculate degradation efficiency η(t) for comparative analysis [99].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential Research Materials for UV-Vis Analysis of Polymers and Biomaterials
| Material/Reagent | Specification | Application Function | Technical Notes |
|---|---|---|---|
| Nanoporous Gold Scaffolds [98] | Pore radius: 34 nm; Thickness: 1-2 μm | Substrate for infiltration studies | Fabricated by dealloying AgAu alloys |
| Poly(2-vinylpyridine) [98] | Molecular weight range: 85-940 kDa | Model polymer for infiltration kinetics | Purified by precipitation before use |
| Polyethylene Automotive Plaques [99] | Injection-molded; 50×50×3 mm | Degradation studies | Standard automotive grade |
| Polypropylene Automotive Plaques [99] | Injection-molded; 50×50×3 mm | Comparative degradation studies | Standard automotive grade |
| pDPP3T Semiconducting Polymer [100] | High purity; film thickness 100-200 nm | Bioelectronic interface studies | Spin-coated from chlorobenzene solutions |
| True-to-Life Nanoplastics [102] | Polystyrene; environmentally relevant | Environmental nanoplastic quantification | Generated from fragmented consumer plastics |
| Quartz Cuvettes [8] | Path length: 1 cm; Spectrosil grade | UV transmission measurements | Required for UV range below 350 nm |
| Microvolume Spectrophotometer [102] | 0.5-2 μL sample requirement | Nanoplastic quantification | Enables sample recovery for additional analyses |
UV-Vis spectroscopy serves as an indispensable analytical technique for characterizing conjugation and degradation in polymeric and biomaterial systems. Through careful monitoring of electronic transitions, researchers can extract quantitative information about molecular structure, aggregation state, infiltration kinetics, and degradation pathways. The technique's versatility extends from fundamental studies of electronic structure to practical applications in automotive durability, bioelectronic interfaces, and environmental nanoplastic quantification.
As demonstrated through the methodologies presented in this guide, successful application of UV-Vis spectroscopy requires thoughtful experimental design, appropriate control experiments, and sophisticated data analysis approaches. When implemented correctly, it provides unique insights into material behavior that complement other analytical techniques and advance our understanding of structure-property relationships in complex polymeric systems.
The continued development of UV-Vis methodologies, including multivariate statistical analysis, kinetic modeling, and correlation with material performance, will further expand its utility in both academic research and industrial applications. By leveraging the fundamental principles of electronic transitions, researchers can address emerging challenges in polymer science and biomaterials engineering through this accessible yet powerful spectroscopic technique.
Ultraviolet-visible (UV-Vis) spectroscopy stands as a fundamental pillar in pharmaceutical analysis, providing critical insights into drug identity, purity, and concentration. This technique operates on the principle that molecules undergo electronic transitions when exposed to specific wavelengths of light, absorbing energy as electrons are promoted from ground state to excited state molecular orbitals [13]. The energy required for these transitions corresponds directly to the wavelength of light absorbed, creating a characteristic spectral fingerprint that enables both qualitative and quantitative analysis of pharmaceutical compounds [8].
The theoretical foundation of UV-Vis spectroscopy revolves around electronic transitions involving π, σ, and n electrons. When molecules contain conjugated π systems, the energy gap between the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) decreases, resulting in absorption at longer wavelengths [4]. This phenomenon explains why compounds with extensive conjugation, such as β-carotene (with 11 conjugated double bonds), absorb visible light and appear colored [13]. Understanding these electronic transitions provides the scientific basis for method selection in pharmaceutical analysis, enabling researchers to predict and interpret spectral data for drug development.
Theoretical Foundations: Electronic Transitions and Spectral Characteristics
Types of Electronic Transitions in Pharmaceutical Compounds
The electronic transitions central to UV-Vis spectroscopy are categorized based on the molecular orbitals involved. The primary transitions relevant to pharmaceutical compounds include:
π→π* transitions: These occur in molecules with conjugated double bonds, where π electrons are excited to π* antibonding orbitals. As conjugation increases, the energy gap (ΔE) decreases, resulting in longer wavelength absorption [13]. For example, ethene absorbs at 170 nm, 1,3-butadiene at 217 nm, and 1,3,5-hexatriene at 258 nm [4].
n→π* transitions: These involve the excitation of non-bonding electrons (lone pairs) to π* orbitals. These transitions typically appear at longer wavelengths than π→π* transitions but with lower absorption intensity [13]. They are particularly relevant for pharmaceutical compounds containing carbonyl, nitro, or other heteroatomic functional groups.
σ→σ* transitions: These require high energy and occur in the far-UV region (<150 nm), making them less practically useful for routine pharmaceutical analysis [4].
The energy relationship governing these transitions follows the equation E = hc/λ, where E is energy, h is Planck's constant, c is the speed of light, and λ is wavelength [4]. This fundamental relationship enables the quantification of electronic transition energies from spectral data.
Instrumentation and Measurement Principles
UV-Vis spectrophotometers operate through several key components that work in concert to measure light absorption [8]:
- Light sources: Tungsten or halogen lamps for visible light (400-700 nm) and deuterium lamps for UV light (200-400 nm)
- Wavelength selection: Monochromators containing diffraction gratings (typically 1200+ grooves/mm) to isolate specific wavelengths
- Sample compartment: Cuvettes (quartz for UV, glass/plastic for visible only) holding the sample and reference solutions
- Detection systems: Photomultiplier tubes (PMTs), photodiodes, or charge-coupled devices (CCDs) to convert transmitted light into electrical signals
The fundamental measurement in UV-Vis spectroscopy follows the Beer-Lambert law: A = εlc, where A is absorbance, ε is the molar absorptivity coefficient (L·molâ»Â¹Â·cmâ»Â¹), l is path length (cm), and c is concentration (mol·Lâ»Â¹) [8]. This relationship enables quantitative analysis, with absorbance values optimally maintained below 1.0 for accurate measurements [8].
Analytical Method Selection Framework
Strategic Approach to Method Selection
Selecting the appropriate analytical technique for pharmaceutical analysis requires systematic evaluation of multiple factors to ensure method suitability, accuracy, and regulatory compliance. The framework begins with comprehensive requirement definition [103]:
- Analyte characteristics: Identity of compounds/elements to be quantified, chemical structure, and specific chromophores
- Concentration levels: Expected analyte concentration and required detection limits
- Sample matrix: Composition of the sample and potential interferents from other analytes
- Accuracy and precision requirements: Acceptable degrees of uncertainty based on intended method use
- Regulatory considerations: Compliance with FDA, EMA, ICH, or other relevant guidelines [104]
Table 1: Analytical Technique Selection Guide Based on Application Requirements
| Analytical Need | Recommended Technique | Typical Pharmaceutical Applications | Key Considerations |
|---|---|---|---|
| Quantitative analysis | UV-Vis Spectroscopy | API potency, content uniformity, dissolution testing | High precision, wide linear range, suitable for quality control |
| Multi-component analysis | HPLC with UV detection | Complex formulations, impurity profiling | Separation capability, specificity, method development time |
| Structural elucidation | NMR, MS | Unknown identification, degradation products | Structural information, sensitivity, instrument availability |
| Trace analysis | LC-MS/MS | Genotoxic impurities, metabolites | Low detection limits, high specificity, cost |
| Volatile compounds | GC-FID/MS | Residual solvents, essential oil components | Volatility requirements, thermal stability |
Following requirement definition, a thorough literature search should be conducted using authoritative sources including pharmacopoeias (USP, EP, BP), CODEX, ASTM, EPA, APHA standards, and indexed scientific journals [103]. This search should prioritize peer-reviewed publications and established standard methods over general internet searches to ensure methodological validity [103].
Practical Considerations for Method Implementation
Beyond theoretical suitability, practical considerations significantly impact method selection success:
- Method complexity: Procedures with numerous stages (extraction, derivatization, multiple dilutions) introduce potential errors and reduce laboratory throughput [103]
- Equipment and reagent availability: Access to required instrumentation and high-quality reagents within the analytical facility
- Personnel expertise: Technical training on instrumentation, method execution, and data interpretation
- Time and cost efficiency: Balance between analysis time, reagent costs, and required throughput
- Validation feasibility: Ability to demonstrate method performance characteristics meet acceptance criteria [104]
The framework emphasizes that the simplest method is not necessarily optimal, nor is the most technically advanced approach always justified. Rather, the selected method should represent the most appropriate balance of scientific rigor, practical feasibility, and regulatory compliance for the specific analytical need [103].
UV-Vis Spectroscopy Method Development Protocol
Systematic Method Development Approach
A structured, step-wise approach to UV-Vis method development ensures robust, reliable analytical methods suitable for pharmaceutical applications:
Step 1: Define analytical objectives - Clearly articulate the method purpose, including the specific attribute to be measured, acceptance criteria, and intended use [104]. This includes understanding critical quality attributes (CQAs) of the drug substance or product.
Step 2: Conduct literature review and method feasibility assessment - Research existing methods for the analyte or structurally similar compounds [104]. Evaluate theoretical suitability based on the analyte's chromophoric properties and expected concentration range.
Step 3: Develop method plan - Outline detailed methodology, instrumentation specifications, and experimental design [104]. Select appropriate reference standards, reagents, and develop preliminary protocols.
Step 4: Method optimization - Systematically adjust parameters including sample preparation, solvent selection, path length, and wavelength selection [104]. For UV-Vis methods, this includes verifying λmax and establishing linear range.
Step 5: Method validation - Execute validation under appropriate conditions (R&D or GLP-compliant) to demonstrate method suitability for intended use [104].
Step 6: Method transfer (if required) - For multi-site manufacturing or clinical trials, conduct formal method transfer with training and documentation [104].
Step 7: Sample analysis - Implement routine analysis under controlled conditions (R&D or cGMP, as appropriate) [104].
Critical Method Validation Parameters
For regulatory acceptance, UV-Vis methods must undergo comprehensive validation demonstrating the following performance characteristics [104]:
- Accuracy: The closeness of test results to the true value, typically established through spike recovery studies
- Precision: The degree of agreement among individual test results, including repeatability and intermediate precision
- Specificity: The ability to measure the analyte accurately in the presence of potential interferents
- Linearity: The ability to obtain test results proportional to analyte concentration within a specified range
- Range: The interval between upper and lower concentration levels with demonstrated accuracy, precision, and linearity
- Limit of Detection (LOD): The lowest amount of analyte that can be detected
- Limit of Quantification (LOQ): The lowest amount of analyte that can be quantified with acceptable accuracy and precision
- Robustness: The capacity to remain unaffected by small, deliberate variations in method parameters
- Ruggedness: The degree of reproducibility under normal operational conditions across different instruments, analysts, and laboratories
Table 2: Typical Validation Acceptance Criteria for UV-Vis Spectrophotometric Methods
| Validation Parameter | Acceptance Criteria | Recommended Approach |
|---|---|---|
| Accuracy | Recovery 98-102% | Spike recovery at 80%, 100%, 120% of target concentration |
| Precision | RSD ≤ 2.0% | Six replicate preparations at 100% target concentration |
| Linearity | R² ≥ 0.998 | Minimum five concentrations across specified range |
| Range | Typically 80-120% of test concentration | Established from linearity data meeting accuracy/precision criteria |
| LOD | Signal-to-noise ratio ≥ 3:1 | Serial dilution until signal-to-noise criteria met |
| LOQ | Signal-to-noise ratio ≥ 10:1, accuracy/precision meet criteria | Serial dilution with accuracy/precision assessment |
| Robustness | No significant impact on results | Deliberate variation of parameters (pH, wavelength, etc.) |
Visualization of Method Selection and Development Processes
Analytical Technique Selection Framework
UV-Vis Method Development Workflow
Electronic Transitions in UV-Vis Spectroscopy
Essential Research Reagents and Materials for Pharmaceutical UV-Vis Analysis
Table 3: Key Research Reagent Solutions for UV-Vis Spectrophotometric Analysis
| Reagent/Material | Specification Requirements | Function in Analysis | Technical Considerations |
|---|---|---|---|
| Reference Standards | Certified purity ≥95% (preferably ≥98%) | Quantitative calibration, method validation | Source from certified suppliers (USP, EP); verify certificate of analysis |
| HPLC-Grade Solvents | UV cutoff below measurement wavelength; low absorbance | Sample preparation, mobile phase | Methanol (UV cutoff 205 nm), Acetonitrile (190 nm), Water (Purified) |
| Buffer Salts | High purity, low UV absorbance | pH control, mobile phase modifier | Phosphate, acetate commonly used; filter through 0.45μm membrane |
| Quartz Cuvettes | Spectrosil grade quartz; matched path length | Sample containment for UV measurement | 1 cm standard path length; ensure cleanliness; check for scratches |
| Mobile Phase Additives | Reagent grade or better | Modification of separation selectivity | Trifluoroacetic acid, ammonium acetate, formic acid |
| Filter Membranes | 0.45μm or 0.22μm pore size | Sample clarification, mobile phase filtration | Nylon, PVDF, or PTFE compatible with solvent system |
| System Suitability Standards | Pharmacopeial standards where available | Verification of instrument performance | USP system suitability references for retention, efficiency, resolution |
The selection of appropriate analytical techniques, particularly UV-Vis spectroscopy, requires systematic evaluation of both theoretical principles and practical constraints. By understanding the fundamental electronic transitions that govern light absorption in pharmaceutical compounds, analysts can make informed decisions about method suitability and optimization. The framework presented integrates theoretical knowledge with practical implementation considerations, providing a structured approach to analytical method selection, development, and validation. As pharmaceutical analysis continues to evolve with advances in instrumentation and computational prediction [105], this foundation in electronic transition theory remains essential for interpreting spectral data and ensuring analytical methods meet the rigorous demands of drug development and quality control.
Conclusion
UV-Vis spectroscopy remains an indispensable, rapid, and cost-effective tool in the researcher's arsenal, fundamentally rooted in the well-understood theory of electronic transitions. Its power for qualitative identification of chromophores and quantitative analysis via the Beer-Lambert law is undeniable. However, its effective application in demanding fields like drug development hinges on a rigorous understanding of its methodological pitfalls, solvent effects, and instrumental limitations. As demonstrated through comparative studies, UV-Vis data gains significant robustness when validated against orthogonal techniques like HPLC and NMR. Future directions point toward increasingly sophisticated spectral analysis, such as Pekarian function fitting, to extract more nuanced electronic and vibrational information from complex molecules, further solidifying its value in the development and quality control of novel biomedical compounds and materials.
References
- 1. 5.5: Ultraviolet and visible spectroscopy [https://chem.libretexts.org/Courses/Brevard_College/CHE_202%3A_Organic_Chemistry_II/05%3A_Structural__Determination_I/5.05%3A_Ultraviolet_and_visible_spectroscopy]
- 2. UV-Vis Absorption Spectroscopy - Theory [https://teaching.shu.ac.uk/hwb/chemistry/tutorials/molspec/uvvisab1.htm]
- 3. Ultraviolet–visible spectroscopy [https://en.wikipedia.org/wiki/Ultraviolet%E2%80%93visible_spectroscopy]
- 4. Introduction To UV-Vis Spectroscopy [https://www.masterorganicchemistry.com/2016/09/16/introduction-to-uv-vis-spectroscopy/]
- 5. UV-Visible Spectroscopy [https://www2.chemistry.msu.edu/faculty/reusch/virttxtjml/spectrpy/uv-vis/spectrum.htm]
- 6. 4.1 Theory of electronic transitions - Spectroscopy [https://fiveable.me/spectroscopy/unit-4/theory-electronic-transitions/study-guide/9SgNHJVlKvKzsC4O]
- 7. Penetration depth and effective sample size ... [https://www.sciencedirect.com/science/article/pii/S0022354925003417]
- 8. UV-Vis Spectroscopy: Principle, Strengths and Limitations ... [https://www.technologynetworks.com/analysis/articles/uv-vis-spectroscopy-principle-strengths-and-limitations-and-applications-349865]
- 9. What Is the Principle of UV-Vis Spectroscopy and Why It ... [https://connect.ssllc.com/learning-center/principle-of-uv-vis-spectroscopy/]
- 10. Interpreting UV Spectra | MCC Organic Chemistry [https://courses.lumenlearning.com/suny-mcc-organicchemistry/chapter/interpreting-uv-spectra/]
- 11. 9.1 UV-visible spectroscopy and electronic transitions [https://fiveable.me/molecular-physics/unit-9/uv-visible-spectroscopy-electronic-transitions/study-guide/NlR9tQyvdmu9422y]
- 12. Molecular electronic transition [https://en.wikipedia.org/wiki/Molecular_electronic_transition]
- 13. 4.4: Ultraviolet and visible spectroscopy [https://chem.libretexts.org/Courses/Oregon_Institute_of_Technology/OIT%3A_CHE_333_-_Organic_Chemistry_III_(Lund)/New_Page/4%3A_Structure_Determination_I-_UV-Vis_and_Infrared_Spectroscopy_Mass_Spectrometry/4.4%3A_Ultraviolet_and_visible_spectroscopy]
- 14. 3.3: Electronic Transitions [https://chem.libretexts.org/Bookshelves/Organic_Chemistry/Introduction_to_Organic_Spectroscopy/03%3A_Conjugated_Compounds_and_Ultraviolet_Spectroscopy/3.03%3A__Electronic_Transitions]
- 15. UV-Vis Spectroscopy: Absorbance of Carbonyls [https://www.masterorganicchemistry.com/2016/09/26/uv-vis-spectroscopy-absorbance-of-carbonyls/]
- 16. Charge Transfer Transition - an overview [https://www.sciencedirect.com/topics/chemistry/charge-transfer-transition]
- 17. UV2. UV-Visible Spectroscopy: Metal Ions. [https://employees.csbsju.edu/cschaller/Principles%20Chem/structure%20determination/UVmetal.htm]
- 18. Charge Transfer Complex between O-Phenylenediamine ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9118382/]
- 19. Electronic Spectra - Ultraviolet and Visible Spectroscopy [https://chem.libretexts.org/Courses/Providence_College/CHM_331_Advanced_Analytical_Chemistry_1/09%3A_Applications_of_Ultraviolet-Visable_Molecular_Absorption_Spectrometry/9.02%3A_Absorbing_Species/9.2.03%3A_Electronic_Spectra_-_Ultraviolet_and_Visible_Spectroscopy_-_Metal_to_Ligand_and_Ligand_to_Metal_Charge_Transfer_Bands]
- 20. Charge migration and charge transfer in molecular systems [https://pmc.ncbi.nlm.nih.gov/articles/PMC5745195/]
- 21. Spectroscopic Investigation of Charge Transfer Interaction ... [https://www.spectroscopyonline.com/view/spectroscopic-investigation-of-charge-transfer-interaction-between-five-antibiotics-depending-on-density-functional-theory]
- 22. Synthesis and Spectroscopic Characterization of Schiff Base ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10882688/]
- 23. Examining Metal Complexes and How They Enhance ... [https://www.spectroscopyonline.com/view/examining-metal-complexes-and-how-they-enhance-detection-techniques]
- 24. 8.6.2: Selection Rules [https://chem.libretexts.org/Courses/Ursinus_College/CHEM322%3A_Inorganic_Chemistry/08%3A_Electronic_Structure_of_Coordination_Complexes/8.06%3A_Tanabe_Sugano_Diagrams/8.6.02%3A_Selection_Rules]
- 25. Laporte rule [https://en.wikipedia.org/wiki/Laporte_rule]
- 26. Derivation of Laporte Rule [https://chem.libretexts.org/Bookshelves/Physical_and_Theoretical_Chemistry_Textbook_Maps/Supplemental_Modules_(Physical_and_Theoretical_Chemistry)/Spectroscopy/Electronic_Spectroscopy/Selection_Rules_for_Electronic_Spectra_of_Transition_Metal_Complexes/Derivation_of_Laporte_Rule]
- 27. La Porte Selection rule.pptx [https://www.slideshare.net/slideshow/la-porte-selection-rulepptx/253067144]
- 28. Spin Selection Rule - (Inorganic Chemistry I) [https://fiveable.me/key-terms/inorganic-chemistry-i/spin-selection-rule]
- 29. Franck Condon Principle | PPTX [https://www.slideshare.net/slideshow/franck-condon-principle/255683481]
- 30. Franck–Condon principle [https://en.wikipedia.org/wiki/Franck%E2%80%93Condon_principle]
- 31. 13.7: The Franck-Condon Principle [https://chem.libretexts.org/Courses/Pacific_Union_College/Quantum_Chemistry/13%3A_Molecular_Spectroscopy/13.07%3A_The_Franck-Condon_Principle]
- 32. light absorption and fate of excitation energy [https://www.life.illinois.edu/govindjee/biochem494/Abs.html]
- 33. Franck-Condon Theory of Quantum Mechanochemistry [https://pubmed.ncbi.nlm.nih.gov/28685567/]
- 34. Forbidden Electron Transfer in the Adiabatic Limit of ... [https://arxiv.org/html/2511.01909v1]
- 35. UV-Visible Spectroscopy [https://www2.chemistry.msu.edu/faculty/reusch/virttxtjml/spectrpy/uv-vis/uvspec.htm]
- 36. Using UV-visible Absorption Spectroscopy [https://chem.libretexts.org/Bookshelves/Physical_and_Theoretical_Chemistry_Textbook_Maps/Supplemental_Modules_(Physical_and_Theoretical_Chemistry)/Spectroscopy/Electronic_Spectroscopy/Electronic_Spectroscopy_Basics/Using_UV-visible_Absorption_Spectroscopy]
- 37. Absorbance Spectroscopy & Spectra Explained [https://www.ossila.com/pages/absorbance-spectroscopy]
- 38. The Beer-Lambert Law [https://chem.libretexts.org/Bookshelves/Physical_and_Theoretical_Chemistry_Textbook_Maps/Supplemental_Modules_(Physical_and_Theoretical_Chemistry)/Spectroscopy/Electronic_Spectroscopy/Electronic_Spectroscopy_Basics/The_Beer-Lambert_Law]
- 39. Beer-Lambert Law | Transmittance & Absorbance [https://www.edinst.com/resource/the-beer-lambert-law/]
- 40. 3.13 Beer-Lambert Law - AP Chem [https://fiveable.me/ap-chem/unit-3/beer-lambert-law/study-guide/smCHzraorVz6qlWW1oeB]
- 41. UV–Vis operando spectroelectrochemistry for (photo) ... [https://www.sciencedirect.com/science/article/pii/S2451910322001636]
- 42. How to Interpret UV-Vis Spectroscopy Results [https://www.azooptics.com/Article.aspx?ArticleID=2768]
- 43. UV/VIS Spectroscopy - an overview [https://www.sciencedirect.com/topics/chemistry/uv-vis-spectroscopy]
- 44. What is a Chromophore? | Functional Groups and Examples [https://www.ossila.com/pages/chromophore]
- 45. 3: UV Spectroscopy [https://chem.libretexts.org/Ancillary_Materials/Worksheets/Spectroscopy_(Worksheets)/03%3A_UV_Spectroscopy]
- 46. UV-Visible Spectroscopy - JASCO Inc [https://jascoinc.com/learning-center/theory/spectroscopy/uv-vis-spectroscopy/]
- 47. Chromophores, Fluorophores: properties and characteristics [https://www.bachem.com/knowledge-center/technical-notes/chromophores-fluorophores-spectral-properties-and-characteristics/]
- 48. Review on qualitative and quantitative of analysis by drugs -visible... uv [https://zenodo.org/records/7982675]
- 49. Derivation of Beer Lambert Law [https://byjus.com/physics/derivation-of-beer-lambert-law/]
- 50. - UV quantification of residual acetone during the... Vis spectroscopic [https://pubmed.ncbi.nlm.nih.gov/30845867/]
- 51. 4.4: UV-Visible Spectroscopy [https://chem.libretexts.org/Bookshelves/Analytical_Chemistry/Physical_Methods_in_Chemistry_and_Nano_Science_(Barron)/04%3A_Chemical_Speciation/4.04%3A_UV-Visible_Spectroscopy]
- 52. NIR-II bioluminescence for in vivo high contrast imaging ... [https://www.nature.com/articles/s41467-020-18051-1]
- 53. Monitoring Reactions Through UV-Visible Spectroscopy [https://www.spectroscopyonline.com/view/monitoring-reactions-through-uv-visible-spectroscopy]
- 54. Comparison of Quantification Using UV-Vis, NMR, and ... [https://www.mdpi.com/1422-0067/26/14/6638]
- 55. Active Ingredient Testing Methods for Spa Formulations [https://www.halecosmeceuticals.com/blog/active-ingredient-testing-methods-spa-formulations]
- 56. 4.9: Ultraviolet Spectroscopy [https://chem.libretexts.org/Courses/University_of_Illinois_Springfield/Introduction_to_Organic_Spectroscopy/4%3A_Conjugated_Compounds_and_Ultraviolet_Spectroscopy/4.09%3A_Ultraviolet_Spectroscopy]
- 57. Molecular Structure And Absorption Spectra - MCAT Content [https://jackwestin.com/resources/mcat-content/molecular-structure-and-absorption-spectra/ultraviolet-region]
- 58. Video: UV – Vis : Molecular Spectroscopy Electronic Transitions [https://www.jove.com/science-education/v/13025/uvvis-spectroscopy-molecular-electronic-transitions]
- 59. in Electronic visible transition UV spectroscopy [https://www.pharmatutor.org/articles/summary-on-electronic-transition-in-uv-visible-spectroscopy]
- 60. Separation, identification, and quantification of active ... [https://www.sciencedirect.com/science/article/pii/S2095177911000670]
- 61. Solvatochromism as a new tool to distinguish structurally ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6440649/]
- 62. Exploring solvatochromism: A comprehensive analysis of ... [https://www.sciencedirect.com/science/article/abs/pii/S0026265X24002789]
- 63. How to Work with Challenging or Limited Genomic ... [https://blog.omni-inc.com/blog/how-to-work-with-challenging-or-limited-genomic-samples-updated-for-2025]
- 64. Sample preparation and cleanup methods for clinical top ... [https://pubmed.ncbi.nlm.nih.gov/40717221/]
- 65. UV-Vis Spectroscopy: A Step in the Light Direction [https://icjs.us/uv-vis-spectroscopy-a-step-in-the-light-direction/]
- 66. What is Stray light and how it is monitored? [https://lab-training.com/what-is-stray-light-and-how-it-is-monitored/]
- 67. Errors in Spectrophotometry and Calibration Procedures to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5293527/]
- 68. Sources of Error in UV-Vis Spectroscopy [https://www.ossila.com/pages/sources-of-error-uv-vis-spectroscopy]
- 69. UV-vis Spectrophotometer Cuvette Selection Guide [https://airekacells.com/blog/cuvette-guide]
- 70. UV vis Spectrophotometer and Fluorescence Cuvettes [https://cotslab.com/guide-uv-vis-spectrophotometer-and-fluorescence-cuvettes/]
- 71. Choosing the Right Cuvettes for UV-VIS Experiments [https://whitebearphotonics.com/blogs/measurement-techniques/uv-vis-cuvette-guide?srsltid=AfmBOooScWgOpsUvX8PlnHBauMLwaPDOFXugBx6WXqZcjpv_pVkg29Nu]
- 72. Stray Light Calibration Standards for Validating UV/VIS ... [https://www.fireflysci.com/stray-light?srsltid=AfmBOoqBBSYbAbfHqjPMQ7A5wPjZYQ1pRREhGfW7rrRzfT-IlxOViiF_]
- 73. Changes in Absorption Spectra - CleanEnergyWIKI [https://cleanenergywiki.org/index.php?title=Changes_in_Absorption_Spectra]
- 74. What Are Chromophores and Chromophoric Shifts? [https://lab-training.com/what-are-chromophores-and-chromophoric-shifts/]
- 75. Chromophore, Auxochrome and different type of shift in UV ... [https://gpatindia.com/chromophore-auxochrome-and-shift/]
- 76. Chromophore [https://en.wikipedia.org/wiki/Chromophore]
- 77. Exploring the Spectrum of Analytical Techniques for ... [https://www.spectroscopyonline.com/view/exploring-the-spectrum-of-analytical-techniques-for-material-characterization]
- 78. Analyzing experimental UV–vis spectra of conjugated ... [https://pubs.rsc.org/en/content/articlehtml/2025/nj/d4nj05537c]
- 79. Spectral Analysis and Reaction Mechanism by QM/MM ... [https://tms.riken.jp/en/research/project-1/]
- 80. UV-Vis Spectroscopy: Principle, Parts, Uses, Limitations [https://microbenotes.com/uv-vis-spectroscopy/]
- 81. UV Spectroscopy Market Size & Share Analysis [https://www.mordorintelligence.com/industry-reports/uv-spectroscopy-market]
- 82. UV & Visible Spectroscopy Insightful Analysis [https://www.archivemarketresearch.com/reports/uv-visible-spectroscopy-504455]
- 83. Advantages and Applications of UV-Vis Analysis [https://www.richmondscientific.com/advantages-and-applications-of-uv-vis-analysis?srsltid=AfmBOopvHIf2kPs2UDTaGfauPG85aM3PYHWqrziMet8k0hp4yeVqF3Om]
- 84. How Chemometrics Revives the UV-Vis Spectroscopy ... [https://www.mdpi.com/2227-9040/11/1/8]
- 85. 2025 Review of Spectroscopic Instrumentation [https://www.spectroscopyonline.com/view/2025-review-of-spectroscopic-instrumentation]
- 86. HPLC Basics | Thermo Fisher Scientific - US [https://www.thermofisher.com/us/en/home/industrial/chromatography/chromatography-learning-center/liquid-chromatography-information/hplc-basics.html]
- 87. Principle of Separation in HPLC [https://www.shimadzu.com/an/service-support/technical-support/analysis-basics/basic/principle_of_separation_in_hplc.html]
- 88. Liquid Chromatographic Peak Purity Assessments in ... [https://www.chromatographyonline.com/view/liquid-chromatographic-peak-purity-assessments-in-forced-degradation-studies---an-industry-perspective]
- 89. Advantages, Limitations, And Future Trends Of High- ... [https://www.alwsci.com/news/advantages-limitations-and-future-trends-of-85237687.html]
- 90. Comparison of high-performance liquid chromatography ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6425260/]
- 91. Comparison between liquid chromatography-UV detection ... [https://pubmed.ncbi.nlm.nih.gov/12350089/]
- 92. How UV-Vis is Evolving in 2025 for Better Lab Efficiency [https://www.richmondscientific.com/how-uv-vis-is-evolving-in-2025-for-better-lab-efficiency?srsltid=AfmBOopqI3pbu7ltckrPE65bSEJu4K68OQle2iivoRUP3IkV5K-Ftro7]
- 93. New HPLC, MS, and CDS Products from 2024–2025 [https://www.chromatographyonline.com/view/new-hplc-ms-and-cds-products-from-2024-2025-a-brief-review]
- 94. A method for validating the accuracy of NMR protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7749147/]
- 95. Validation of a Quantitative Proton Nuclear Magnetic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7023380/]
- 96. 1 H NMR spectra modeling for predicting the acid ... [https://www.sciencedirect.com/science/article/abs/pii/S0308814625011525]
- 97. The study of aggregation dynamics of conjugated polymer ... [https://www.sciencedirect.com/science/article/pii/S2405844021007416]
- 98. Investigating polymer infiltration kinetics in nanoporous metal scaffolds... [https://pubs.rsc.org/en/content/articlelanding/2025/sm/d5sm00423c]
- 99. Multi-Statistical Pragmatic Framework to Study UV Exposure ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12608747/]
- 100. Simple surface treatment of conjugated polymers for ... [https://chemrxiv.org/engage/chemrxiv/article-details/678a49d26dde43c908ef3ab0]
- 101. Polystyrene and UVC Sterilization Tested with ... [https://www.spectroscopyonline.com/view/polystyrene-and-uvc-sterilization-tested-with-spectroscopy-and-luminescence-tools]
- 102. Quantitative evaluation of true-to-life nanoplastics using UV ... [https://pubs.rsc.org/en/content/articlehtml/2025/en/d5en00502g]
- 103. Important Considerations in Selection of Method for ... [https://lab-training.com/important-considerations-selection-method-required-analysis/]
- 104. A Step-by-Step Guide to Analytical Method Development ... [https://emerypharma.com/blog/a-step-by-step-guide-to-analytical-method-development-and-validation/]
- 105. Comparative dataset of experimental and computational ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6895184/]