Advanced Strategies for Calibration Drift Correction in Field Spectrometers
This article provides a comprehensive guide for researchers and drug development professionals on managing calibration drift in field spectrometers.
Advanced Strategies for Calibration Drift Correction in Field Spectrometers
Abstract
This article provides a comprehensive guide for researchers and drug development professionals on managing calibration drift in field spectrometers. It covers the fundamental causes of drift, including environmental factors and instrumental aging, and explores a range of correction methodologies from traditional standardization to advanced machine learning algorithms. The content offers practical troubleshooting advice, compares the performance of different correction techniques through real-world case studies, and delivers actionable validation protocols to ensure data integrity in long-term biomedical and clinical research studies.
Understanding Calibration Drift: Sources and Impacts on Data Integrity
Defining Calibration Drift and Its Critical Importance in Field Spectroscopy
Definition and Causes of Calibration Drift
What is calibration drift? Calibration drift is the slow, often monotonic change in the response of a measurement instrument over time, leading to a gradual loss of measurement accuracy. It is a key concern in field spectroscopy, where it can cause skewed readings and increase measurement uncertainty, potentially compromising data integrity and research outcomes [1].
What are the primary causes of calibration drift? The causes are multifaceted and can include:
- Environmental Factors: Sudden changes in temperature or humidity are common causes of drift [1]. Deep-sea environments, combining low temperatures and high pressure, present particular challenges [2].
- Instrument Aging and Use: The natural aging of electronic or mechanical components, along with frequent use, accelerates the need for recalibration [1].
- Physical Stress: Exposure to harsh conditions, mechanical shock, or vibration can induce calibration errors [1].
- Optical Component Issues: Dirty windows or lenses in front of fiber optics or light pipes can cause analysis to drift [3].
- Component Degradation: Aging light sources (e.g., calibration lamp bulb degradation) and detectors can alter the system's response [4].
Quantitative Data on Calibration Drift
The following table summarizes documented drift rates and impacts from various spectroscopic and sensor applications.
Table 1: Documented Calibration Drift Rates and Impacts
| Instrument/Sensor Type | Observed Drift Rate / Magnitude | Primary Cause / Context | Impact on Measurement |
|---|---|---|---|
| Iridium-192 Brachytherapy Source | +0.15% per year (post-2018) [5] | Update of primary metrology standards [5] | Dosimetric discrepancies in medical radiotherapy [5] |
| Quartz Resonance Pressure Sensor | Increased drift with deployment depth [2] | Deep-sea environment (low temperature, high pressure) [2] | Accumulated depth and positioning errors [2] |
| Optical π-FBG Pressure Sensor | Annual drift reduced to < ±0.002 MPa after correction [2] | Sensor aging in deep-sea conditions [2] | High-precision depth measurement for underwater navigation [2] |
| Calibration Light Source | Output change: ~0.1%/hr at 350 nm; ~0.02%/hr at 900 nm [4] | Natural bulb degradation over time [4] | Introduces uncertainty in radiometric calibration [4] |
Troubleshooting FAQs for Field Spectrometers
Q1: My spectral analysis results are inconsistent between measurements of the same sample. What should I check?
- Step 1: Clean optical windows. Dirty windows on the fiber optic or direct light pipe are a common cause of drift and poor results [3]. Clean them according to the manufacturer's guidelines.
- Step 2: Verify calibration. Recalibrate your spectrometer. It is recommended to calibrate before starting a job and at least once daily [6].
- Step 3: Check the vacuum pump (if applicable). In optical emission spectrometers, a malfunctioning vacuum pump can cause low-intensity loss for elements like Carbon, Phosphorus, and Sulfur, leading to incorrect values [3]. Listen for unusual pump noises and check for oil leaks [3].
- Step 4: Ensure proper sample preparation. Ensure samples are not contaminated during handling (e.g., by skin oils) and are properly prepared (e.g., reground) before analysis [3].
Q2: How can I tell if my spectrophotometer's color measurements are drifting?
- Symptom: Customers may reject shipments due to color inaccuracies, or you may identify color issues during quality control checks [6].
- Prevention: Regular calibration and maintenance are critical. A device should be calibrated each time you start a job and at least once a day. The longer you go without calibration, the harder it is for the device to correct itself [6].
- Verification: For fleets of instruments, use a tool like NetProfiler to check wavelength errors and validate that all devices are measuring consistently on a monthly basis [6].
Q3: What is the recommended long-term maintenance schedule for a field spectrometer?
- Daily/Per Job: Perform a full instrument calibration [6].
- Weekly/Monthly: Clean the instrument's optical components, such as the aperture and white reference tile, following the user manual. Frequency depends on the operating environment (e.g., daily in a factory, monthly in a clean lab) [6].
- Annually: Seek factory ISO certification. Annual certification verifies measurement accuracy, provides NIST traceability, and is often required for regulatory compliance [6].
Advanced Drift Correction Methodologies
For long-term research projects, advanced correction methods are essential for data integrity. These often involve algorithmic correction and specialized experimental design.
Algorithmic Correction for Long-Term Studies
In techniques like Gas Chromatography-Mass Spectrometry (GC-MS), signal drift over extended periods (e.g., 155 days) can be corrected using quality control (QC) samples and machine learning [7].
- Workflow: A virtual QC sample is established from all QC measurements. Correction factors for each chemical component are calculated based on its peak area and a function of batch number and injection order. Algorithms like Random Forest (RF) are then used to model and correct the drift in sample data [7].
- Performance: Research shows that the Random Forest algorithm provides the most stable and reliable correction for long-term, highly variable data, outperforming Spline Interpolation and Support Vector Regression [7].
In-Situ Drift Correction with Reference Probes
A powerful hardware-based method for field sensors involves using a reference probe deployed alongside the primary sensor [2].
- Principle: The reference probe is designed to be insensitive to the primary analyte (e.g., pressure) but remains exposed to the same environmental conditions (e.g., temperature) that cause drift. The drift coefficients are extracted from the reference probe and used to correct the primary sensor's output [2].
- Experimental Validation: This method has been validated in simulated deep-sea environments, where it reduced the annual drift of an optical pressure sensor to less than ±0.002 MPa. Time-series forecasting with an ARIMA model suggested a calibration period not exceeding six months for long-term deployment [2].
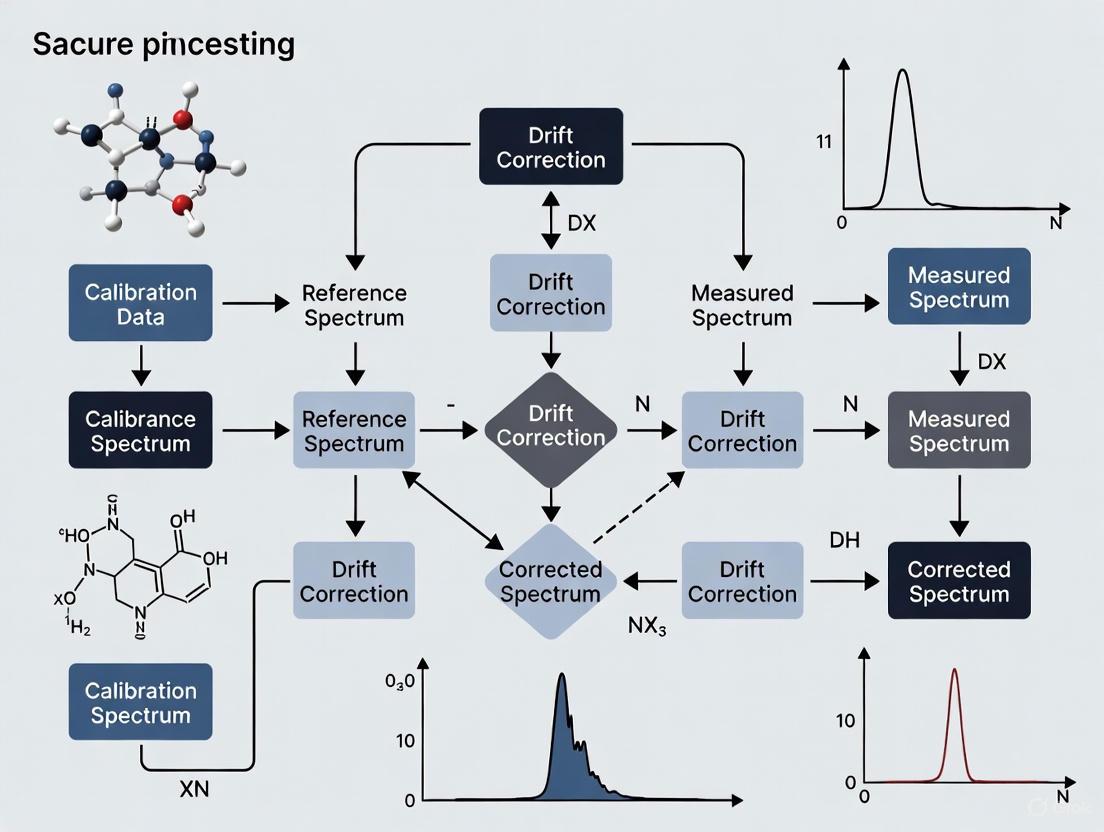
The diagram below illustrates the logical workflow and relationship between components in a reference probe correction system.
In-Situ Drift Correction with a Reference Probe
The Scientist's Toolkit: Key Reagents & Materials
Table 2: Essential Materials for Drift Monitoring and Correction
| Item Name | Function / Purpose |
|---|---|
| NIST-Traceable Calibration Light Source | Provides a known spectral output to calibrate the spectrometer's radiometric response. Requires periodic recalibration due to inherent output degradation (e.g., ~0.1%/hr at 350 nm) [4]. |
| Emission Line Sources (Hg, Ar, Xe, etc.) | Used for precise wavelength calibration. The distinct atomic emission lines allow for accurate characterization and correction of the wavelength axis across the spectrometer's range [4]. |
| Non-Absorbing Reference Matrix (KBr, KCl) | Essential in techniques like DRIFTS for diluting powdered samples to minimize spectral artefacts and provide a consistent scattering environment for quantitative analysis [8]. |
| Stray Light Filters (e.g., Holmium Oxide) | Used to measure and characterize stray light within the spectrometer, which is critical for manufacturing quality control and long-term performance tracking [4]. |
| Virtual QC Sample | A computational construct in chromatographic analysis, created from pooled Quality Control sample data. Serves as a meta-reference for normalizing test samples and correcting for long-term signal drift using algorithms [7]. |
Instrumental drift is a pervasive challenge in analytical measurements, undermining the long-term reliability and accuracy of field spectrometers and other chemical sensors. It refers to the gradual, undesired change in a sensor's quantitative response over time, even when measuring a constant standard. For researchers and scientists, effectively troubleshooting drift is paramount for ensuring data integrity. This guide provides a structured framework to diagnose and address the primary sources of instrumental drift, categorized into environmental influences and internal hardware factors.
The first step in diagnosis is to identify the likely source of the drift. The table below contrasts common symptoms and examples of these two primary drift types.
| Characteristic | Environmental Variation Drift | Hardware/Instrumental Drift |
|---|---|---|
| Primary Cause | Changes in the external measurement environment or sample matrix [9] [10] | Physical aging and changes within the instrument itself [9] [10] |
| Example Factors | Ambient temperature, humidity, pressure, and interfering chemical species [10] | Sensor aging (e.g., catalyst degradation, membrane fouling), component wear, and source intensity decay [11] [10] [12] |
| Typical Signal Behavior | Often correlated with measured environmental parameters; can be reversible or cyclical [9] | Generally exhibits a monotonic trend (e.g., constant shift or gradual slope) over time [11] |
Troubleshooting FAQs and Guides
How can I determine if drift is due to environmental changes or the instrument itself?
Objective: To isolate the root cause of observed drift in sensor measurements.
Experimental Protocol:
- Step 1: Collect Concurrent Data: During your measurements, systematically record not only the sensor's output but also relevant environmental variables such as ambient temperature, humidity, and atmospheric pressure [9].
- Step 2: Analyze with Reference Data: Compare your sensor's readings to those from a co-located, high-accuracy reference instrument, if available [9].
- Step 3: Apply Probabilistic Modeling: Use statistical techniques to decouple the influences. Importance sampling can be employed to re-weight data and isolate the instrumental drift component from the environmental variation [9]. Plot your sensor error against the logged environmental variables to identify correlations.
- Interpretation: A strong correlation between the sensor error and an environmental parameter (like temperature) points to environmental variation as a primary cause. A steady, uncorrelated change in the signal suggests inherent instrumental drift [9].
What are the established methods for correcting instrumental drift?
Objective: To apply mathematical or procedural corrections to compensate for instrumental drift.
Experimental Protocol:
- Step 1: Regular Drift Monitoring: Use a stable, known reference material (a "drift monitor") not used in the original calibration. Measure this monitor at regular intervals throughout an analytical run [12].
- Step 2: Calculate a Correction Factor: For each monitoring session, calculate a drift correction factor, ( R(t) ). This can be based on a known isotope ratio in mass spectrometry or a standard signal intensity in spectroscopy [11] [12]. ( R(t) = \frac{I!R(t)}{I!RT} ) Where ( I!R(t) ) is the measured ratio at time ( t ), and ( I!RT ) is the known true value.
- Step 3: Apply the Correction: Use the calculated ( R(t) ) to correct your sample measurements. This can be done via an interpolating polynomial fitted to the monitor's drift over time or by using the monitor as an internal standard for ratio-based correction [11].
Our calibration model has become unstable. How can we update it without a full recalibration?
Objective: To update a multivariate calibration model using a minimal set of new standard samples.
Experimental Protocol:
- Step 1: Standardization Subset Selection: Identify a small, representative set of standard samples (the "standardization subset") [10].
- Step 2: Measure Under New Conditions: Measure this subset under the new conditions where the model is failing (e.g., on a new instrument, or after significant drift has occurred).
- Step 3: Model Transfer: Establish a mathematical relationship (e.g., using Piecewise Direct Standardization (PDS)) between the responses of the original (master) instrument and the drifted (child) instrument using the standardization subset data [13] [10].
- Step 4: Apply Transformation: Use this relationship to transform new measurements from the child instrument to the scale of the master instrument, allowing the original calibration model to be applied accurately [13].
Instrumental Drift Detection and Correction Workflow
The following diagram illustrates a generalizable workflow for detecting and responding to calibration drift, adaptable to various instrumental platforms.
Research Reagent Solutions for Drift Management
The following table details key materials and their functions in monitoring and correcting for instrumental drift.
| Reagent/Material | Primary Function | Field of Application |
|---|---|---|
| Drift Monitors (e.g., fused glass discs) | Stable reference materials for regular instrument monitoring to quantify and correct for intensity drift of the X-ray tube [12]. | X-Ray Fluorescence (XRF) Spectrometry |
| Certified Reference Materials (CRMs) | High-accuracy standards used for initial calibration and periodic validation of instrument performance. | All Spectroscopic Methods |
| Internal Standard Solution | A known compound added to all samples and standards to correct for signal fluctuations and instrument drift [10]. | Chromatography, Mass Spectrometry |
| Isotopically Labeled Standards | The most accurate internal standard for isotope dilution methods, correcting for sample preparation losses and instrument drift [10]. | Isotope Ratio Mass Spectrometry |
| Standardization Subset | A small set of stable standards used to transfer a calibration model from a master to a child instrument without full recalibration [13] [10]. | Multivariate Calibration (e.g., NIR, E-nose) |
Frequently Asked Questions (FAQs)
Q1: What are the primary sources of spectral variability between instruments? The main sources of inter-instrument variability are wavelength alignment errors, spectral resolution and bandwidth differences, and detector and noise variability. These hardware-induced spectral variations cause models developed on one instrument to fail when applied to data from other spectrometers [14].
Q2: How does environmental humidity affect spectrometer measurements? Water vapor causes substantial spectral interference, leading to significant biases in measurements. For methane isotopic composition (δ¹³CH₄) measurements, humidity-induced biases can be corrected using empirical correction functions (quadratic for ¹²CH₄ and ¹³CH₄, linear for δ¹³CH₄) established over a water vapor range of 0.15–4.0% [15].
Q3: What is long-term instrumental drift and how can it be corrected? Long-term drift refers to gradual changes in instrument response over extended periods. In GC-MS instruments over 155 days, effective correction uses quality control (QC) samples and algorithms like Random Forest, which provided the most stable and reliable correction model for highly variable data [7]. For CO₂ sensors, drifts producing biases up to 27.9 ppm over 2 years can be corrected via linear interpolation [16].
Q4: Can calibration models be transferred between different instruments? Yes, but this requires specific standardization techniques. Methods like Direct Standardization (DS), Piecewise Direct Standardization (PDS), and External Parameter Orthogonalization (EPO) can map spectral domains between master and slave instruments, though each method has limitations regarding linearity assumptions and computational requirements [14].
Troubleshooting Guides
Inaccurate Analysis Results
| Symptom | Possible Cause | Solution |
|---|---|---|
| Significant variation between tests on same sample | Improper calibration | Recalibrate using proper sequence; analyze standard sample 5x consecutively; RSD should not exceed 5% [3] |
| Consistent low readings for carbon, phosphorus, sulfur | Vacuum pump malfunction | Check pump for noise, overheating, or oil leaks; service or replace pump [3] |
| Drifting results or frequent need for recalibration | Dirty optical windows | Clean windows in front of fiber optic and in direct light pipe [3] |
| Inconsistent readings between replicates | Sample degradation or improper handling | Ensure sample is light-stable; minimize time between measurements; use same cuvette orientation [17] |
Signal and Noise Issues
| Symptom | Possible Cause | Solution |
|---|---|---|
| Unstable or drifting readings | Insufficient instrument warm-up | Allow spectrophotometer to warm up for 15-30 minutes before use [17] |
| Low light intensity or signal error | Dirty optics or misaligned cuvette | Clean sample cuvette; check for debris in light path; ensure proper cuvette alignment [18] |
| High noise rate and dark current | Spacer design in MRPC detectors | Pad spacers outperform fishline spacers by reducing electrical creepage effects in high-rate conditions [19] |
| Negative absorbance readings | Blank solution dirtier than sample | Use exact same cuvette for blank and sample measurements; ensure proper blank solution [17] |
Environmental Impact on Sensor Accuracy
| Sensor Type | Environmental Factor | Impact on Accuracy | Correction Method | Post-Correction Accuracy |
|---|---|---|---|---|
| Cavity Ring-Down Spectrometer [15] | Water vapor (0.15-4.0%) | Substantial biases in δ¹³CH₄ | Empirical humidity correction | Stable & accurate across conditions |
| NDIR CO₂ Sensor (SENSE-IAP) [16] | Temperature, humidity | RMSE: 5.9 ± 1.2 ppm | Multivariate linear regression | RMSE: 1.6 ± 0.5 ppm |
| SenseAir K30 CO₂ Sensor [16] | Environmental factors | ±30 ppm ±3% of reading | Environmental correction | RMSE: 1.7-4.3 ppm |
Long-Term Drift Performance and Correction
| Instrument Type | Drift Magnitude | Time Period | Correction Algorithm | Performance After Correction |
|---|---|---|---|---|
| GC-MS [7] | Large fluctuations | 155 days | Random Forest (RF) | Most stable and reliable correction |
| GC-MS [7] | Large fluctuations | 155 days | Support Vector Regression (SVR) | Tendency to over-fit and over-correct |
| GC-MS [7] | Large fluctuations | 155 days | Spline Interpolation (SC) | Least stable performance |
| NDIR CO₂ Sensor [16] | Up to 27.9 ppm bias | 2 years | Linear interpolation | RMSE reduced to 2.4 ± 0.2 ppm |
Calibration Transfer Technique Comparison
| Method | Principle | Advantages | Limitations |
|---|---|---|---|
| Direct Standardization (DS) [14] | Linear transformation between instruments | Simple, efficient with paired samples | Vulnerable to local nonlinear distortions |
| Piecewise Direct Standardization (PDS) [14] | Localized linear transformations | Handles local nonlinearities better than DS | Computationally intensive; can overfit noise |
| External Parameter Orthogonalization (EPO) [14] | Removes non-chemical variability | Works without paired sample sets | Requires proper estimation of orthogonal subspace |
Experimental Protocols
Humidity-Induced Bias Correction for Methane Isotopic Measurements
Purpose: To establish and apply correction functions for accurate δ¹³CH₄ measurements in both dry and humid air [15].
Materials:
- Cavity ring-down spectrometer
- Nafion dryer for dry air measurements
- Humidity control system
- Reference gas standards
Procedure:
- Perform laboratory experiments across water vapor range of 0.15–4.0%
- Establish empirical correction functions:
- Quadratic corrections for ¹²CH₄ and ¹³CH₄ concentrations
- Linear correction for δ¹³CH₄ values
- Apply correction functions to field measurements in both dried air (SORPES site) and humid air (Jurong site)
- Compare performance of delta-based calibration versus isotopologue-specific calibration
- Validate with reference measurements
Key Findings: Isotopologue-specific calibration coupled with explicit water vapor correction delivered stable and accurate δ¹³CH₄ measurements across all conditions, while delta-based calibration showed significant biases in humid air correlated with 1/CH₄ [15].
Long-Term Drift Correction Using Quality Control Samples
Purpose: To correct instrumental data drift in GC-MS measurements over 155 days using quality control samples and multiple algorithms [7].
Materials:
- GC-MS instrument
- Quality control samples (pooled)
- Reference standards
- Computational resources for algorithm implementation
Procedure:
- Conduct 20 repeated tests on six commercial tobacco products over 155 days
- Establish "virtual QC sample" by incorporating chromatographic peaks from all 20 QC results
- Classify sample components into three categories:
- Category 1: Present in both QC and sample
- Category 2: In sample only but within retention time tolerance
- Category 3: In sample only, outside retention time tolerance
- Calculate correction factors for component k in i-th measurement: yᵢ,ₖ = Xᵢ,ₖ/XT,ₖ where Xᵢ,ₖ is peak area, XT,ₖ is median peak area across all measurements
- Apply three correction algorithms:
- Spline Interpolation Correction (SC)
- Support Vector Regression (SVR)
- Random Forest (RF)
- Evaluate performance using principal component analysis and standard deviation analysis
Key Findings: Random Forest algorithm provided the most stable and reliable correction model for long-term, highly variable data, while SC showed the least stability and SVR tended to over-fit [7].
Low-Cost Sensor Drift Evaluation and Correction
Purpose: To evaluate and correct long-term drifts in low-cost CO₂ sensors over 30 months of field deployment [16].
Materials:
- SENSE-IAP sensor units (SenseAir K30)
- Picarro reference analyzer (G2401)
- Environmental monitoring equipment
- Calibration gases
Procedure:
- Perform co-located observations using LCS units alongside Picarro reference analyzer
- Develop environmental correction system using multivariate linear regression
- Monitor long-term drifts and seasonal drift cycles
- Apply linear interpolation for drift correction
- Evaluate correction effectiveness by comparing with reference measurements
- Determine optimal calibration frequency
Key Findings: Environmental correction reduced RMSE from 5.9 ± 1.2 to 1.6 ± 0.5 ppm. Long-term drifts produced biases up to 27.9 ppm over 2 years, but linear interpolation reduced 30-month RMSE to 2.4 ± 0.2 ppm. Recommended calibration frequency: within 3 months, not exceeding 6 months [16].
Workflow Visualization
Instrument Calibration Transfer Workflow
Long-Term Drift Correction Process
Research Reagent Solutions
| Reagent/Material | Function | Application Context |
|---|---|---|
| Pooled Quality Control (QC) Samples [7] | Establish correction dataset for long-term drift | GC-MS measurements over extended periods |
| Nafion Dryer [15] | Remove water vapor from air samples | Humidity control for spectral measurements |
| Certified Reference Standards [18] | Instrument calibration and validation | Spectrophotometer accuracy verification |
| Low-Resistive Glass [19] | Improve rate capability in MRPC detectors | High-rate timing detectors for physics experiments |
| Virtual QC Sample [7] | Meta-reference for normalization | Long-term GC-MS studies with changing components |
| SenseAir K30 Sensor [16] | Low-cost CO₂ monitoring | Urban emission network deployment |
The Real-world Impact of Uncorrected Drift on Long-term Study Reliability
Your Troubleshooting Guide to Instrument Drift
Instrument drift is a gradual change in an instrument's measurement output over time, even when measuring the same sample. In long-term studies, uncorrected drift can compromise data integrity, leading to inaccurate results and unreliable conclusions [20] [21]. This guide helps you identify, correct, and prevent drift in your research.
Frequently Asked Questions (FAQs)
1. What are the real-world consequences of uncorrected drift in my research? Uncorrected drift can severely impact your study's reliability. In a clinical study using an electronic nose for disease detection, sensor drift had a more profound effect on the results than the actual clinical disease state, threatening the diagnostic algorithm's validity [21]. In reading research, drift can move eye fixations from one word or line to another, leading to the misidentification of eye-movement effects and incorrect research findings [22]. In spectroscopy, drift directly reduces the accuracy and repeatability of your elemental analysis [20] [12].
2. I use an XRF Spectrometer. How can I check for drift? The standard method is to use a dedicated drift monitor [20] [12]. These are stable, glass-fused discs with a known elemental composition.
- Procedure: Regularly measure the drift monitor using your standard method. A change in the measured count rate compared to its established reference value indicates instrument drift.
- Key Point: Drift monitors are for monitoring instrument stability and performing drift correction; they are not Certified Reference Materials (CRMs) for calibration [20] [12].
3. My spectrophotometer's color measurements are inconsistent. Is this drift? Yes, spectrophotometers are susceptible to color drift due to factors like temperature changes, aging light sources, and photo detector degradation [6]. To confirm and correct:
- Daily Calibration: Calibrate your instrument at the start of every job and at least once daily [6].
- Regular Cleaning: Clean the aperture and white tile regularly according to the manufacturer's guidelines to prevent contaminants from causing errors [6].
- Annual Certification: For absolute confidence, have your device factory-certified annually to ensure it meets original performance specifications [6].
4. How can I correct for drift in my datasets after collection? Post-acquisition software correction is a powerful approach. Methods vary by field:
- Eye-Tracking: Expert manual correction is considered highly accurate. Automated algorithms also exist and are improving, with novices performing on par with the best algorithms [22].
- Biomolecular NMR Spectroscopy: The SAFR (Simultaneous Acquisition of a Frequency Reference) method can be used. An auxiliary 1D spectrum is acquired with each scan to monitor and correct for magnetic field drift during the experiment [23].
- Electronic Nose Data: Machine learning techniques like Kernel Transformation (DCKT) and Deep-learning Neural Networks can be employed to correct for sensor drift [21].
Quantitative Impact of Drift
The tables below summarize data on drift magnitude and correction effectiveness across different instruments.
Table 1: Documented Drift Magnitude in Various Instruments
| Instrument Type | Observed Drift | Impact & Context |
|---|---|---|
| Hydraulic Pressure Gauge [24] | ~0.02% of output (after 140 days at 100 MPa) | Significant for long-term monitoring of stable pressures, such as in seafloor crustal movement detection. |
| Electronic Nose (Cyranose 320) [21] | Sensor response drift over 4 days | Drift effect was more profound than clinical features, directly impacting diagnostic algorithm accuracy. |
| XRF Spectrometer [20] | Range of inconsistent results for the same substance | Compromises reliability and accuracy of elemental composition data. |
Table 2: Effectiveness of Different Drift Correction Methods
| Correction Method | Field of Application | Reported Outcome / Benefit |
|---|---|---|
| Drift Monitor Use [20] [12] | XRF Spectrometry | Ensures long-term stability and optimal performance; reduces need for frequent full recalibrations. |
| Manual Correction [22] | Eye-Tracking (Reading) | Expert correction is significantly more accurate than automated algorithms. |
| SAFR Method [23] | Biomolecular Solid-State NMR | Corrects non-linear field drifts, allowing high-quality spectra recording even during field changes of ~0.1 ppm/h. |
| Logistic Regression Correction [21] | Electronic Nose Diagnostics | Improved accuracy for differentiating patient groups (Accuracy: 0.68). |
Experimental Protocols for Drift Assessment & Correction
Protocol 1: Routine Drift Monitoring for an XRF Spectrometer This protocol uses a drift monitor to correct instrument readings [20] [12].
- Acquire a Drift Monitor: Obtain a drift monitor disc (e.g., Ausmon) with a composition similar to your samples (e.g., cement, nickel, silicates).
- Establish a Baseline: When the instrument is known to be well-calibrated, measure the drift monitor multiple times to establish a stable, reference count rate.
- Implement Routine Checks: Before each measurement session or at regular intervals, measure the same drift monitor under identical conditions.
- Calculate and Apply Correction: Compute a correction factor based on the ratio of the current reading to the reference reading. Apply this factor to subsequent sample measurements to correct for the instrument's drift.
Protocol 2: The SAFR Method for NMR Spectrometry This protocol corrects for magnetic field drift during long NMR experiments [23].
- Modify Pulse Sequence: Add a small-flip-angle reference pulse (e.g., 1°) at the beginning of each scan in your multi-dimensional experiment.
- Simultaneous Acquisition: Acquire the auxiliary 1D reference spectrum immediately after the reference pulse. This spectrum, typically of a solvent signal, acts as a navigator for the magnetic field's state.
- Reconstruct Field Evolution: Process the raw data to track the frequency of the solvent signal's maximum across all scans. This reconstructs the temporal evolution of the magnetic field drift.
- Apply Data Correction: Use the reconstructed field drift data to apply a phase correction to the raw FID (Free Induction Decay) data of the main experiment in both the direct and indirect dimensions.
The Researcher's Toolkit: Essential Materials for Drift Management
Table 3: Key Research Reagents and Materials for Drift Control
| Item | Function | Example & Notes |
|---|---|---|
| Drift Monitors [20] [12] | Stable reference materials to monitor and correct for instrument drift in spectrometers. | XRF Scientific Ausmon discs (e.g., for cement, iron ore). Note: These are not Certified Reference Materials (CRMs). |
| Certified Reference Materials (CRMs) | To verify instrument accuracy and perform full calibration. | NIST-traceable standards. Used for initial calibration and periodic validation. |
| Non-Absorbing Matrix [8] | A spectral reference material for DRIFTS to enhance signal quality and minimize artefacts. | KBr (Potassium Bromide) or KCl for mid-IR measurements; diamond powder for robust applications. |
| Stable Polystyrene Standard [13] | A reference standard for verifying wavelength/wavenumber accuracy in spectrophotometers. | Highly crystalline polystyrene filter (e.g., 1mm thickness). |
Workflow Diagram for Managing Instrument Drift
The following diagram illustrates a logical workflow for diagnosing and addressing instrument drift in your experiments.
Correction Methodologies: From Traditional Standardization to Machine Learning
FAQs and Troubleshooting Guides
Frequently Asked Questions
1. What is the primary goal of Direct Standardization (DS) and External Parameter Orthogonalization (EPO) in field spectroscopy?
The main objective of both DS and EPO is to remove the interfering effects of external parameters, such as variable soil moisture content, from vis–NIR spectra. This correction is necessary to enable accurate predictions of soil properties, like Soil Organic Carbon (SOC), using calibration models developed on air-dried spectral libraries [25].
2. When should I consider using these techniques?
These techniques are particularly valuable when you aim to use a spectral library built under controlled, lab-based conditions (e.g., on air-dried soils) to predict properties from spectra collected in the field, where variable conditions like moisture content can severely degrade prediction accuracy [25].
3. How do I know if moisture is affecting my spectral data?
A clear indicator is a decrease in the accuracy of your SOC prediction models. If predictions become less reliable when using field-moist spectra compared to air-dried spectra, moisture is likely a contributing factor [25].
4. Can these techniques be applied to instruments other than vis–NIR spectrometers?
Yes, the principle of correcting for external parameters is universal. For instance, the EPO method was originally derived from a technique called EPO-PLS, which was developed for temperature-independent measurement of sugar content in intact fruits [25].
Troubleshooting Common Issues
Issue: Corrected spectra do not improve prediction model accuracy.
- Potential Cause 1: The calibration set used for the standardization does not adequately represent the moisture variations encountered in your field samples.
- Solution: Ensure the calibration samples used to develop the DS or EPO transformation cover a wide and representative range of moisture conditions [25].
- Potential Cause 2: The model drift may be too severe or non-linear for a linear correction method to handle effectively.
- Solution: Investigate more advanced non-linear calibration techniques or ensure that the pre-launch or pre-deployment calibration of the instrument is robust, as initial calibration stability is critical for long-term data accuracy [26] [27].
Issue: Results are inconsistent after applying correction techniques.
- Potential Cause: Inconsistent sample preparation can introduce new variations that the model cannot separate from the target signal.
- Solution: Standardize sample handling protocols meticulously. For soil samples, ensure consistent grinding and avoid contaminants like oils from skin or quenching media, as these can lead to unstable and inconsistent results [3].
Quantitative Comparison of Techniques
The following table summarizes the performance of two moisture correction methods based on an experimental study aiming to predict Soil Organic Carbon (SOC) [25].
Table 1: Performance Comparison of Soil Moisture Correction Methods for SOC Prediction
| Method | Key Principle | Required Calibration Set | Reported R² | Reported RMSE |
|---|---|---|---|---|
| Direct Standardization (DS) | Transforms spectra from moist conditions to match their appearance under dry conditions. | A set of samples scanned under both moist and dry conditions. | 0.84 | 0.45% |
| External Parameter Orthogonalization (EPO) | Projects spectra into a space orthogonal to the directions of variation caused by moisture. | A set of samples scanned under both moist and dry conditions. | 0.87 | 0.41% |
Detailed Experimental Protocols
Protocol 1: Applying External Parameter Orthogonalization (EPO)
This protocol outlines the steps to remove the effect of water from vis–NIR spectra using the EPO method [25].
- Calibration Set Preparation: Collect a subset of soil samples (
n=50in the cited study) that represent the expected range of soil types and moisture contents. - Spectral Acquisition under Different Conditions: Scan each sample in the calibration set twice:
- Condition A (Field-Moist): Scan the samples at their natural, field-moist state.
- Condition B (Oven-Dried): Oven-dry the same samples at 105°C for 24 hours and then scan them again. These spectra represent the "reference" or target state.
- Calculate Spectral Difference Matrix: For each sample, compute a difference spectrum:
Difference = Spectrum_moist - Spectrum_dry. - Perform Decomposition: Apply a principal component analysis (PCA) to the matrix containing all the difference spectra.
- Develop EPO Transformation: Use the first few principal components (which represent the main directions of moisture-induced variation) to create a projection matrix
P. - Apply Correction: Transform all future field-moist spectra (
X_moist) to their corrected, dry-like form using the equation:X_corrected = X_moist * P.
Protocol 2: Applying Direct Standardization (DS)
This protocol describes the process for correcting spectra using the DS method [25].
- Calibration Set Preparation: Follow the same steps 1 and 2 as in the EPO protocol to obtain paired spectral data from moist and dry conditions.
- Establish Transformation Matrix: Instead of using a difference matrix, the DS algorithm calculates a transformation matrix
Fthat directly maps the entire moist spectrum to its corresponding dry spectrum. - Apply Transformation: For any new field-moist spectrum, the corrected spectrum is calculated as:
X_corrected = X_moist * F.
Method Workflow and Relationships
The following diagram illustrates the logical workflow and relationship between the different standardization techniques and their application.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 2: Key Materials for Spectroscopic Calibration and Drift Correction Experiments
| Item Name | Function / Explanation |
|---|---|
| Calibration Samples | A set of well-characterized samples (e.g., various soil types, alloy standards) used to develop the transformation between different instrument states or environmental conditions [25] [26]. |
| NIST-Traceable Standards | Reference materials with known, certified properties used for primary instrument calibration to ensure data accuracy and traceability to international standards [27]. |
| High-Purity Argon Gas | In Laser-Induced Breakdown Spectroscopy (LIBS), argon is used to create a controlled atmosphere for the plasma. Contaminated argon leads to inconsistent and unstable analytical results [3]. |
| Certified Wavelength Calibration Source | A source containing known emission lines (e.g., from elements like Ti, Fe) used to calibrate the wavelength axis of a spectrometer, which is critical for correct peak assignment, especially when dealing with instrumental drift [26]. |
Leveraging Quality Control Samples for Long-term Drift Correction
This technical support guide provides researchers and scientists with practical methodologies for detecting and correcting long-term instrumental drift in field spectrometers and analytical instruments using Quality Control (QC) samples.
Frequently Asked Questions
1. What is long-term instrumental drift, and why is it a problem? Long-term drift is a gradual change in an instrument's measurement signal over extended periods. It is caused by factors such as component aging (e.g., light source degradation in spectrometers), environmental changes, and routine maintenance operations like column replacement or ion source cleaning [16] [28]. This drift introduces bias and reduces the reliability of quantitative data, making it difficult to compare results from experiments conducted over weeks or months, which is critical for long-term studies and ensuring product stability [28] [29].
2. How can Quality Control (QC) samples correct for this drift? QC samples, typically a pooled mixture representative of your test samples, are measured at regular intervals throughout an experimental timeline. The variation in the measured response of the QC components over time is used to model the instrumental drift. This model generates correction factors that can be applied to your actual experimental data, normalizing them to a stable baseline and compensating for the drift [28] [29].
3. What is the recommended frequency for running QC samples? The optimal frequency depends on the instrument's stability and the required data precision. Research in gas chromatography-mass spectrometry (GC-MS) has successfully used 20 QC measurements over a 155-day period to model drift [28] [29]. For CO2 sensors, maintaining calibration (a form of QC) every 3 months, and not exceeding 6 months, is recommended to ensure accuracy within 5 ppm [16]. A good practice is to analyze a QC sample at the beginning of each batch and after every few experimental samples.
4. My sample contains compounds not present in the QC pool. Can they still be corrected? Yes, strategies exist for this scenario. Components can be categorized based on their presence in the QC, and different correction approaches are used [28]:
- Category 1: Components present in both QC and sample are directly corrected using their own drift model.
- Category 2: Components in the sample not matched in the QC, but with a similar retention time (in chromatography), can be corrected using the model from the adjacent QC peak.
- Category 3: Components in the sample completely absent from the QC can be normalized using an average correction coefficient derived from all QC components [28].
Troubleshooting Guide: QC-Based Drift Correction
| Problem | Possible Causes | Recommended Solutions |
|---|---|---|
| Poor Drift Correction | QC samples not representative of test samples; incorrect correction algorithm [28]. | Ensure the pooled QC contains all (or most) chemicals found in your test samples. For complex data, use robust algorithms like Random Forest instead of simpler interpolation [28]. |
| High Variation in Corrected Data | QC measurements are too infrequent to capture drift pattern; instrument has high short-term noise [28] [17]. | Increase the frequency of QC sample analysis. Ensure the instrument is properly warmed up (15-30 mins) and maintained (clean optics, stable environment) before analysis [17]. |
| Drift Model Fails After Maintenance | Major hardware changes (e.g., new lamp, column) alter the instrument's response, resetting the drift curve [28]. | Consider the post-maintenance data as a new "batch." Re-establish the baseline by running a series of QC samples to build a new drift model for the new batch [28]. |
| Inconsistent Correction Across Components | Different chemical components or sensor responses drift at different rates and are influenced by different factors [16] [28]. | Build a separate correction model, f(p, t), for each key component or response, rather than using a global model for all data [28]. |
Experimental Protocol: Establishing a QC-Based Drift Correction System
The following workflow details the steps for implementing a drift correction procedure, based on methodologies successfully applied in GC-MS studies [28] [29].
Step-by-Step Methodology
Step 1: Preparation of Pooled Quality Control (QC) Sample
- Procedure: Combine equal aliquots from all test samples to be analyzed over the long-term study to create a homogeneous pooled QC sample. This ensures the QC contains a representative profile of all target analytes [28].
- Storage: Divide the pooled QC into single-use aliquots and store under conditions that preserve stability (e.g., -80 °C) to prevent composition changes from affecting the drift model.
Step 2: Establishing the Analysis Schedule
- Procedure: At the start of the experiment, analyze the QC sample 5-10 times to establish a robust baseline and estimate initial variability. Thereafter, analyze a single QC sample at regular intervals interspersed with the test samples. A practical approach is to run a QC after every 4-10 test samples or at the start and end of each analytical batch [28] [30].
- Documentation: Record two key parameters for every analysis (QC and test samples):
- Batch Number (
p): An integer incremented each time the instrument is shut down and restarted or undergoes major tuning. - Injection Order Number (
t): An integer representing the sequence of analysis within a batch [28].
- Batch Number (
Step 3: Data Collection and Calculation of Correction Factors
- Procedure: Collect all data, including the peak areas or instrument responses for each component in the QC samples over time.
- Calculation: For each component
kin thenQC samples, calculate its true value (X_T,k) as the median of all its measured peak areas. Then, compute the correction factor (y_i,k) for that component in thei-thQC measurement using the formula [28]:- Formula:
y_i,k = X_T,k / X_i,k - Where
X_i,kis the raw peak area for componentkin thei-thQC measurement.
- Formula:
Step 4: Modeling Drift and Correcting Data
- Procedure: Use the calculated correction factors
{y_i,k}as the target variable and the batch (p) and injection order (t) numbers as input features to train a drift correction model,f_k(p, t) - Algorithm Selection: The following table compares algorithms evaluated for this purpose in a 155-day GC-MS study [28]:
| Algorithm | Description | Pros / Cons | Best For |
|---|---|---|---|
| Spline Interpolation (SC) | Uses segmented polynomials for interpolation. | Simpler approach, but showed the least stability with sparse data [28]. | Preliminary analysis. |
| Support Vector Regression (SVR) | A variant of SVM for continuous function prediction. | Can over-fit and over-correct with highly variable data [28]. | Datasets with low noise. |
| Random Forest (RF) | An ensemble learning method using multiple decision trees. | Most stable and reliable for long-term, highly variable data [28]. | Complex, long-term datasets. |
- Application: For a given test sample analyzed at batch
pand injection ordert, the corrected valuex'_s,kfor componentkis calculated as [28]:- Formula:
x'_s,k = x_s,k * f_k(p, t) - Where
x_s,kis the raw measurement in the test sample.
- Formula:
Step 5: Validation of the Correction Procedure
- Procedure: Validate the effectiveness of the drift correction by performing Principal Component Analysis (PCA) on the QC data before and after correction. Successful correction will show tight clustering of all QC samples in the PCA scores plot, indicating reduced variance over time [28]. Additionally, the standard deviation of each component across the corrected QC samples should be significantly reduced.
Quantitative Data from Research Studies
The following tables summarize empirical findings on drift magnitude and correction efficacy from published research.
Table 1: Drift Magnitude and Correction in CO2 Sensor Networks
This data is from a 30-month field evaluation of low-cost CO2 sensors [16].
| Metric | Value Before Correction | Value After Correction |
|---|---|---|
| Daily RMSE | 5.9 ± 1.2 ppm | 1.6 ± 0.5 ppm |
| Long-term Drift Bias | Up to 27.9 ppm over 2 years | 2.4 ± 0.2 ppm (with interpolation) |
| Seasonal Drift RMSE | Up to 25 ppm after 6 months | Maintained within 1-3 ppm daily |
Table 2: Performance of Correction Algorithms in GC-MS
This data is from a study comparing three algorithms over 155 days and 20 QC runs [28].
| Algorithm | Stability & Reliability | Suitability |
|---|---|---|
| Random Forest (RF) | Most stable and reliable | Highly variable, long-term data |
| Support Vector Regression (SVR) | Less stable, tends to over-fit | Datasets with lower variation |
| Spline Interpolation (SC) | Least stable with sparse data | -- |
The Scientist's Toolkit: Essential Research Reagents & Materials
| Item | Function in Drift Correction |
|---|---|
| Pooled QC Sample | A composite of all test samples; serves as the stable, representative material used to model instrumental drift over time [28]. |
| Certified Reference Materials (CRMs) | Materials with certified stability and traceable values; can be used as a superior QC sample or to validate the accuracy of the correction method [31]. |
| Internal Standards | Compounds added to both QC and test samples; correct for variations during sample preparation and analysis, complementing the inter-sample drift correction [28]. |
| Particle Size Standards | Suspensions of monodisperse particles; used for the calibration and validation of particle sizing/counting instruments, ensuring that part of the measurement system is standardized [32]. |
| Stable Control Samples | For non-chromatographic instruments like field spectrometers, a stable, homogeneous physical standard (e.g., a colored tile or sealed gas cell) acts as the QC sample to track instrument drift [16] [33]. |
Frequently Asked Questions
Q1: What is calibration drift and why is it a problem for field spectrometers? Calibration drift refers to the gradual change in an instrument's measurement accuracy over time. For field spectrometers, this is a critical problem because it compromises the reliability and portability of data. Inter-instrument variability, caused by factors like wavelength alignment errors, spectral resolution differences, and detector noise variability, means a model developed on one spectrometer often fails on another, even of the same model. This drift can lead to inaccurate chemical quantification and requires correction for trustworthy analytical results [14].
Q2: When should I choose SVR over Random Forest for drift correction, and vice versa? The choice depends on your data's characteristics and the desired outcome. Support Vector Regression (SVR) is particularly powerful for capturing complex, non-linear relationships, especially when you have a clear theoretical margin of tolerance (epsilon) for errors [34] [35]. Random Forest often demonstrates superior stability with highly variable data and can better maintain calibration across different probability ranges, making it less prone to overfitting in these scenarios [36] [7]. For long-term, highly variable datasets, one study found Random Forest to provide the most stable correction [7].
Q3: My SVR model is overfitting the drift in my training data. How can I improve it? Overfitting in SVR is often a result of hyperparameter misconfiguration. You can address it by:
- Reducing the model complexity: Decrease the value of the
Cparameter. A lowerCvalue creates a softer margin, allowing for more errors and a simpler model [37] [35]. - Adjusting the kernel: If using an RBF kernel, increase the
gammaparameter. A highergammamakes the model more sensitive to individual data points, so reducing it can help smooth the decision boundary [35]. - Increasing epsilon: A larger
epsilonvalue widens the tolerance margin, making the model less sensitive to small fluctuations and noise in the training data [35].
Q4: How can I preprocess my spectral data for effective drift correction with these algorithms? Proper preprocessing is vital. Key steps include:
- Feature Scaling: Always normalize or standardize your features before using SVR, as it is sensitive to the scale of data [34] [35]. Algorithms like
StandardScalerin Python are commonly used. - Quality Control (QC) Samples: Regularly measure pooled QC samples throughout your data acquisition period. These samples serve as a reference to model and correct for instrumental drift over time [7].
- Parameterizing Drift: Represent the drift by incorporating parameters like batch number (to mark instrument power cycles or maintenance) and injection order number (to track sequence within a batch) as input features for your correction model [7].
Troubleshooting Guides
Issue: Poor Performance After Transferring Model to a New Spectrometer
Description: A calibration model that performs well on the original ("master") instrument produces inaccurate predictions when applied to a new ("slave") spectrometer or the same instrument after a period of time [14].
Diagnosis: This is a classic symptom of inter-instrument variability or calibration drift. The source of the spectral distortion must be identified.
Solution:
- Diagnose the Source of Variability:
- Wavelength Misalignment: Check for small shifts in the wavelength axis. This can distort the alignment of absorbance bands with the original model's regression vector [14].
- Resolution Differences: Differences in spectral resolution or bandwidth can broaden or narrow spectral peaks, altering their shape [14].
- Detector Noise: Changes in signal-to-noise ratio between instruments can introduce systematic errors [14].
- Apply Calibration Transfer Techniques:
- Direct Standardization (DS): Assumes a global linear transformation between the slave and master spectra. It is simple but may not handle local non-linearities well [14].
- Piecewise Direct Standardization (PDS): An improvement over DS that applies localized linear transformations across the spectrum, offering better handling of local distortions [14].
Issue: Random Forest Model Fails to Correct Drift for Low-Abundance Compounds
Description: The drift correction model works for major constituents but fails for trace-level components in the sample.
Diagnosis: The pooled Quality Control (QC) sample may not adequately represent low-abundance compounds, or the signal for these compounds is too weak for the model to learn a reliable correction function [7].
Solution:
- Enhance QC Samples: Ensure your pooled QC sample contains a representative amount of the low-abundance compounds. This may require spiking the QC with these analytes.
- Implement Advanced Correction Strategies: For components not present in the QC samples, use alternative normalization methods. One approach is to use the average correction factor derived from all reliable QC components. Another is to use the correction factor from a chromatographic peak adjacent to the target compound, assuming the drift behavior is similar [7].
- Data Filtering: As a last resort, consider filtering out highly unstable spectral features that cannot be reliably corrected, as this can improve the overall statistical power for the remaining, stable features [38].
Experimental Protocol: Correcting GC-MS Long-Term Drift
The following table summarizes a published methodology for correcting long-term instrumental drift in Gas Chromatography-Mass Spectrometry (GC-MS) data, which is directly applicable to spectrometer data [7].
- Objective: To establish a reliable protocol for correcting long-term signal drift in GC-MS data using machine learning algorithms.
- Summary: The study involved repeated tests on smoke from six commercial tobacco products over 155 days using a GC-MS instrument. Twenty pooled QC samples were measured throughout this period to model the drift.
| Protocol Aspect | Details |
|---|---|
| Experimental Duration | 155 days [7] |
| Quality Control (QC) | 20 pooled QC samples were analyzed periodically [7] |
| Data Parameters | Batch number (to mark instrument power cycles) and injection order number (sequence within a batch) were recorded for each measurement [7] |
| Algorithms Tested | Spline Interpolation (SC), Support Vector Regression (SVR), Random Forest (RF) [7] |
| Key Innovation | Creation of a "virtual QC sample" from all QC results for normalization and accounting for batch/injection order effects [7] |
| Performance Outcome | Random Forest provided the most stable and reliable correction for long-term, highly variable data [7] |
The workflow for this experiment is outlined below.
The Scientist's Toolkit: Essential Reagents & Materials
The following table lists key materials and computational tools required for establishing a drift correction protocol for field spectrometers, based on the cited research.
| Item | Function / Description |
|---|---|
| Pooled Quality Control (QC) Sample | A composite sample containing aliquots from all samples to be analyzed. It serves as a reference for modeling instrumental drift over time [7]. |
| Internal Standards (IS) | A set of known compounds added to samples to correct for variations in sample preparation and instrument response. Useful when QC samples lack some sample components [7]. |
| StandardScaler (or similar) | A software tool for feature scaling. It is critical for SVR performance to normalize all input features to the same scale [34] [35]. |
| scikit-learn Library (Python) | A machine learning library that provides implementations for both Support Vector Regression (SVR) and Random Forest algorithms [37] [39]. |
| R-based Scripts | Custom scripts, such as those used for nonlinear retention time correction and peak alignment in metabolomics data, can be adapted for spectral drift correction [38]. |
Hyperparameter Tuning for Optimal Performance
The performance of SVR and Random Forest is highly dependent on their hyperparameters. The table below provides a guide for tuning them in the context of drift correction.
| Algorithm | Key Hyperparameters | Function & Tuning Guidance |
|---|---|---|
| Support Vector Regression (SVR) | Kernel (e.g., rbf, linear) [37] [39] |
Determines the model's ability to capture non-linearity. RBF is a good default for complex drift patterns [37] [35]. |
| C (Regularization) [37] [35] | Controls the trade-off between a smooth model and fitting the training data perfectly. A lower C can prevent overfitting to noisy drift data. |
|
| Gamma (for RBF kernel) [35] | Defines the influence of a single training example. Lower values create a smoother model, which can be more robust. | |
| Epsilon (ε) [35] | Defines the margin of error within which no penalty is given. A larger epsilon creates a wider, more tolerant margin. | |
| Random Forest | n_estimators [36] | The number of trees in the forest. A higher number generally improves performance but increases computational cost. |
| max_features [36] | The number of features to consider for the best split. It controls the randomness and strength of the trees. | |
| max_depth [36] | The maximum depth of the trees. Limiting depth helps prevent overfitting. |
The process of building and applying a drift correction model can be visualized as follows.
Implementing Real-time Drift Monitoring with Probability Distribution Functions (PDFs)
Troubleshooting Guides
Guide 1: Troubleshooting Particle Filter Estimator Divergence
Problem: The state estimates from your Particle Filter (PF) become inaccurate and do not converge, even with a sufficient number of particles.
| Symptoms | Potential Causes | Corrective Actions |
|---|---|---|
| Rapidly decreasing effective particle count [40] | Excessive process noise; model mismatch with true system dynamics. | Tune the process noise covariance matrix (Q); validate and refine your system model f(x(t-1), u(t-1), v(t-1)) [40]. |
| High variance in particle weights, with most weights near zero [40] | Severe sensor fault or drift distorting the measurement update. | Implement sensor fault detection; inspect the consistency between model predictions and observations [40]. |
| State distribution becomes overly dispersed and fails to track the true state [40] | Insufficient particle diversity leading to sample impoverishment. | Increase the number of particles; consider implementing a more advanced resampling algorithm [40]. |
Guide 2: Resolving Inadequate Drift Correction Performance
Problem: The drift correction algorithm is running, but the corrected data still shows significant residual drift or introduces new artifacts.
| Symptoms | Potential Causes | Corrective Actions |
|---|---|---|
| Corrected signal shows high-frequency noise or distortions. | The drift model is overfitting to short-term variations in the signal rather than the long-term drift. | Apply a smoothing constraint or reduce the complexity of the drift model. For PDF-based methods, analyze the correlation structure of states for inconsistencies [40]. |
| The calibration model becomes invalid after sensor replacement. | The calibration was not successfully transferred to the new sensor set. | Apply calibration transfer techniques like Direct Standardization (DS) or Piecewise Direct Standardization (PDS) using a small set of standardization samples [41]. |
| Corrected data violates fundamental physical or chemical relationships (e.g., Kramers-Kronig relations in EIS) [42]. | The drift correction method is applied to a system with a fundamentally changing impedance, which it cannot correct. | Do not rely on drift correction. Re-evaluate measurement stability; shorten measurement time or investigate system non-stationarity [42]. |
Frequently Asked Questions (FAQs)
FAQ 1: What is the fundamental difference between implicit and explicit drift correction methods?
- Implicit Correction Methods (ICM): These methods expand the calibration model by incorporating new data collected under drifting conditions. They add drift as a new source of variance in the model itself. This requires periodic reference measurements to update the model [43].
- Explicit Correction Methods (ECM): These methods explicitly model the drift component, often as a direction or a subspace in the data. The calibration model is then made orthogonal or invariant to this estimated drift space. ECM can be more efficient as it directly targets and removes the identified drift [43].
FAQ 2: When is it inappropriate to use algorithmic drift correction?
Algorithmic drift correction is not a universal solution. It is typically inappropriate when:
- The underlying system impedance or properties are fundamentally changing during the measurement (non-stationary behavior) [42].
- The drift is highly non-linear and abrupt, and your model (e.g., a simple polynomial) cannot capture its dynamics [44].
- There is a physical sensor failure or a major change in the sensor array that requires hardware intervention or full recalibration [41].
FAQ 3: How can I monitor the health and performance of my Particle Filter in real-time?
You can monitor several key indicators derived from the estimated state PDF [40]:
- Distribution Heterogeneity: Use entropy or the ratio of effective-to-total particles to ensure the particle set does not collapse.
- Correlation Structure: Track the correlation coefficients between state variables over time. A sudden breakdown in the expected correlation pattern can indicate a fault or model mismatch.
- Prediction-Observation Consistency: Continuously check if the actual measurements are consistent with the distribution of predictions made by your model particles.
FAQ 4: What are the main strategies for maintaining a calibration model over time?
The three primary strategies are [41]:
- Drift Correction: Modeling the sensor drift directly and applying a corrective transform to new data.
- Calibration Standardization: Correcting new data to make it appear as if it was generated under the original calibration conditions, using a small set of standard samples.
- Calibration Update: Incorporating new data from the drifting conditions into the original calibration model to expand its validity.
Experimental Protocols
Protocol: Implementing Particle Filter-Based Drift Monitoring
This protocol outlines the steps for setting up a real-time drift monitor using Particle Filters, as derived from fault diagnostics research [40].
Objective: To detect and diagnose sensor drift by analyzing the probability distribution of system states estimated by a Particle Filter.
Materials:
- A defined state-space model of your system (Eq. 1 and 2 in [40]).
- A stream of real-time sensor data.
- Computing environment capable of running Particle Filter algorithms.
Methodology:
- Initialization: Define your state-transition function
fand measurement functionh. Initialize the PF withN_sparticles, drawing from your initial state distribution. - State Estimation Loop: For each new time step
t:- Prediction: Propagate each particle forward using the state-transition function
f, adding process noisev(t-1). - Update: Upon receiving a new measurement
y(t), calculate the likelihood for each particle and update its weight accordingly. - Resampling: If the effective number of particles falls below a threshold, perform resampling to avoid weight degeneracy.
- Output: Obtain the estimated state PDF from the weighted particles.
- Prediction: Propagate each particle forward using the state-transition function
- Drift Monitoring: Calculate the following metrics from the estimated PDF in real-time:
- Entropy: Monitor the dispersion of the state distribution.
- Effective Particle Ratio:
N_eff = 1 / sum(w_i^2), wherew_iare the particle weights. A low ratio indicates potential problems. - Residuals: Analyze the difference between the actual measurement and the weighted mean of the predicted measurements from the particles.
Diagram: Particle Filter Monitoring Workflow
The Scientist's Toolkit
Research Reagent Solutions
| Item | Function in Drift Monitoring |
|---|---|
| Particle Filter (PF) | A Monte Carlo-based state estimator that approximates the posterior Probability Distribution Function (PDF) of system states, which is essential for tracking non-linear and non-Gaussian processes [40]. |
| Standardization Samples | A small, stable set of physical samples with known properties. Used to establish a relationship between instrument responses under different conditions (e.g., before and after drift) for calibration transfer [41]. |
| State-Space Model | A mathematical model consisting of state-transition and measurement equations. It defines the expected system dynamics and is the core model used by the Particle Filter for prediction [40]. |
| Orthogonal Signal Correction (OSC) | A data pre-processing technique that filters out variation in the sensor data that is orthogonal (unrelated) to the target analyte, often used to remove structured noise like drift [41]. |
| Temporal Convolutional Neural Network (TCNN) | A lightweight deep learning model suitable for embedded deployment. It can learn to model and correct for complex drift patterns directly from temporal sensor data [44]. |
Key Metrics for PDF Analysis
The following table summarizes quantitative metrics for monitoring the health of your PDF-based drift monitor, derived from particle filter diagnostics [40].
| Metric | Formula/Description | Interpretation |
|---|---|---|
| Effective Sample Size ((N_{eff})) | ( N{eff} = \frac{1}{\sum{i=1}^{Ns} (wi^{(t)})^2} ) | A low ( N_{eff} ) indicates weight degeneracy and a loss of particle diversity. |
| Shannon Entropy ((H)) | ( H(X) = - \sum{i=1}^{Ns} wi \log(wi) ) (for discrete weights) | Measures the uncertainty or dispersion of the state distribution. A sudden change can signal a fault. |
| State Correlation ((\rho)) | Pearson correlation coefficient between different state variables, calculated from the particle cloud. | A breakdown in the expected correlation structure can reveal sensor faults or actuator failures. |
Optimizing Performance: Best Practices and Proactive Maintenance
Establishing Optimal Calibration Frequency and Schedules
FAQs on Calibration Frequency and Scheduling
1. What factors determine the optimal calibration frequency for a field spectrometer? The optimal frequency is not a fixed interval but is determined by several factors, including the instrument's inherent stability, the criticality of the measurements, and the operating environment. Instruments in harsh conditions with fluctuating temperatures or humidity, or those used for compliance-critical measurements, require more frequent calibration. A key strategy is to analyze historical calibration drift trends; consistent deviation in analyzer readings over time is a primary indicator that the current schedule is insufficient [45] [46].
2. How can I extend the time between calibrations? Proactive maintenance and advanced data processing can help extend calibration validity. This includes ensuring stable operating conditions (e.g., using temperature control), performing regular instrument health checks (e.g., inspecting light sources and optics), and employing mathematical drift correction or calibration update techniques. These methods use a small set of standard samples to model and correct for drift, reducing the need for full recalibration [41].
3. Our lab has multiple spectrometers. How can we manage calibration efficiently? Implementing a strategic calibration transfer framework can significantly reduce the experimental burden. This involves building a robust calibration model on a primary instrument and then transferring it to other similar instruments using a minimal set of standardization samples. Research shows that methods like ridge regression with Orthogonal Signal Correction (OSC) preprocessing are particularly effective for this, maintaining accuracy while reducing calibration runs by 30-50% [47].
4. What are the immediate signs that my spectrometer needs recalibration? The most common signs include inconsistent readings or drift (where results change systematically over time without a change in the sample), unexpected baseline shifts, and failed calibration gas checks. For quantitative analysis, a growing bias in prediction errors is a clear red flag [48] [49] [45].
Troubleshooting Common Calibration Drift Issues
| Issue | Possible Cause | Corrective Action | |
|---|---|---|---|
| Inconsistent Readings/Drift | [48] [45] | Aging light source (e.g., deuterium lamp), temperature fluctuations, sensor aging, dirty optics. | Replace lamps per manufacturer's schedule; allow instrument warm-up; track drift trends; clean sample cuvettes and optics with lint-free cloth. |
| Failed Calibration Gas Checks | [45] | Expired or contaminated calibration gas; leaks in gas delivery lines; incorrect gas flow rates. | Use NIST-traceable, in-date gases; perform leak checks on all connections; verify gas flow rates (e.g., 1-2 L/min) with a calibrated flow meter. |
| Low Signal Intensity | [49] | Scratched or dirty sample cuvette; misaligned cuvette; debris in the light path. | Inspect and clean the cuvette; ensure proper alignment in the sample holder; check for obstructions in the optical path. |
| Unexpected Baseline Shifts | [49] | Residual sample contamination in the flow cell; electronic drift; need for baseline correction. | Perform a full baseline correction or recalibration; thoroughly clean the flow cell between samples. |
| Software/Data Handling Errors | [45] | Miscommunication between analyzer and data system; unsynchronized system clocks; misconfigured calibration logic. | Audit Data Acquisition System (DAHS) programming; synchronize clocks between all devices; review and test alarm thresholds. |
Experimental Protocol: Automated Calibration and Drift Monitoring
This protocol, inspired by automated systems like ATLAS (Automated Transient Learning for Applied Sensors), outlines a method for efficient calibration model building and continuous drift assessment [50].
Objective: To rapidly develop a multivariate calibration model (e.g., PLS or Ridge Regression) for a field spectrometer and establish a framework for ongoing drift monitoring with minimal manual intervention.
Materials and Reagents:
- Stock Solutions: Certified standard solutions for all analytes of interest.
- Automated Fluid Handling System: A system with pneumatic or syringe pumps and in-line flow meters for precise control.
- Flow Cell: A suitable flow cell compatible with your spectrometer.
- Data Analysis Software: Software capable of multivariate modeling (e.g., Python with SciKit Learn, Unscrambler).
Procedure:
- Experimental Design: Generate an optimal experimental design (e.g., D- or I-optimal) to define the combination of analytes and concentrations for the calibration set. This minimizes the number of required samples while maximizing information [50] [47].
- System Setup: Connect stock solutions to the automated fluid handling system. Program the system to deliver the specified concentrations from the experimental design to the flow cell positioned in the spectrometer.
- Data Acquisition: The automated system cycles through the calibration concentrations. The spectrometer collects spectral data continuously. Flow rates and spectral data are logged simultaneously.
- Model Building: Post-run, extract spectral data from stable periods at each concentration level. Use these spectra and the known concentrations to build a multivariate calibration model (e.g., PLSR or Ridge Regression).
- Drift Monitoring Implementation: Program the system to periodically run a small set of validation standards (e.g., daily or weekly). Use the predictions for these standards to monitor for drift. Establish control limits; if predictions exceed these limits, it triggers a calibration review or update.
Advanced Drift Correction Methodologies
For integration into a research thesis, the following advanced concepts are key. The table below summarizes computational and mathematical approaches to drift correction beyond simple recalibration.
| Method | Principle | Application Context | |
|---|---|---|---|
| Orthogonal Signal Correction (OSC) | [47] [41] | Removes signal components orthogonal to the analyte concentration that are often associated with drift. | Used as a preprocessing step before PLS or Ridge Regression to enhance model robustness against drift. |
| Differentiable Programming | [51] | Uses automatic differentiation to optimize calibration parameters by minimizing the difference between measured data and a target probability distribution. | High-precision X-ray spectroscopy; can be adapted to improve energy scale calibration and reduce systematic uncertainty. |
| Component Correction (e.g., PCA, CCA) | [41] | Models the direction of drift in the sensor response space using a series of measurements, then corrects new data by projecting out this direction. | Electronic noses/tongues; used for classification tasks when sensor drift is the main concern. |
| Calibration Transfer (DS, PDS) | [47] [41] | Standardizes signals between a master and slave instrument (or across time on the same instrument) using a small set of standard samples, enabling model sharing. | Pharmaceutical PAT, multisensor systems; allows calibration models to remain valid when instruments are replaced or drift. |
Workflow for Differentiable Programming Calibration
The following diagram illustrates the advanced calibration method that uses differentiable programming to enhance precision, which is particularly relevant for managing drift in complex spectroscopic data [51].
The Scientist's Toolkit: Key Research Reagents & Materials
| Item | Function in Calibration Research |
|---|---|
| Certified Reference Materials (CRMs) | Provide a traceable, accurate standard for establishing the fundamental calibration curve and validating instrument accuracy [45] [46]. |
| NIST-Traceable Calibration Gases | Essential for gas-phase spectroscopy and CEM systems to ensure the known concentration of analytes delivered during calibration [45]. |
| Drift Monitors / Stable Solid Standards | Specialized, stable materials (e.g., Ausmon drift monitors) used to track instrument stability over time and trigger corrective actions before significant drift occurs [46]. |
| Optical Alignment Sets | Tools for maintaining optimal light path configuration, as misalignment is a common source of signal degradation and drift [48]. |
| Automated Fluidics System | Enables rapid, precise, and reproducible delivery of liquid standards for high-throughput calibration model development and validation [50]. |
Environmental Control and Sample Preparation to Minimize Drift
FAQs on Drift Causes and Prevention
What is calibration drift and why is it a problem? Calibration drift is the slow, unwanted change in the response of a measurement instrument over time, leading to a gradual loss of accuracy. This can result in skewed readings, increased measurement risk, and can compromise the longevity of your equipment. In research, unaddressed drift directly compromises data integrity and the reliability of scientific conclusions [1].
What are the most common causes of drift in field spectrometers? The primary causes can be categorized as follows:
- Environmental Factors: Sudden changes in ambient temperature or humidity are major contributors. Temperature fluctuations can cause materials in the instrument to expand or contract, leading to mechanical drift [1] [52] [17].
- Instrument Handling: Mechanical shock from dropping the device or exposure to vibrations can misalign sensitive optical components [1] [52].
- Sample Preparation Issues: Using dirty or scratched cuvettes, improper blanking, or having air bubbles in your sample can cause significant light scattering and unstable readings [17].
- Natural Degradation: All instruments will experience a slow decline in performance over time and with use due to the aging of components like lamps and electronic parts [1].
How can I stabilize my spectrometer's baseline? Achieving a stable baseline starts with a consistent routine.
- Warm-Up: Always allow the instrument's lamp to warm up for at least 15-30 minutes before use to let the light source stabilize [17] [53].
- Proper Blanking: Use the exact same solvent or buffer for your blank that your sample is dissolved in, and ensure the cuvette is clean [17].
- Stable Environment: Place the spectrometer on a sturdy, level surface away from drafts, vibrations, and significant temperature fluctuations [17] [48].
Troubleshooting Guide: Unstable or Drifting Readings
The table below outlines common symptoms, their likely causes, and recommended solutions.
| Problem | Possible Cause | Recommended Solution |
|---|---|---|
| Unstable / Drifting Readings | Instrument lamp not stabilized [17]. | Allow a 30-minute warm-up period before taking measurements [53]. |
| Air bubbles in the sample [17]. | Gently tap the cuvette to dislodge bubbles; re-prepare sample if persistent. | |
| Sample is too concentrated [17]. | Dilute the sample to bring its absorbance into the optimal range (0.1–1.0 AU). | |
| Unstable environment (vibrations, temperature drafts) [1] [52]. | Place instrument on a vibration-isolation platform; shield from drafts and HVAC vents. | |
| Cuvette is dirty, scratched, or has fingerprints [17] [48]. | Clean with lint-free cloth; handle by frosted sides; replace if scratched. | |
| Cannot Set 100% Transmittance (Fails to Blank) | Light source (lamp) is near end of its life [17]. | Check lamp usage hours; replace old or failing lamps per manufacturer's schedule [53]. |
| Contamination on internal optics [48]. | Schedule professional servicing to clean or realign internal components. | |
| Negative Absorbance Readings | Blank was "dirtier" than the sample (e.g., different cuvette used) [17]. | Use the exact same cuvette for both blank and sample measurements. |
| The blank cuvette was smudged during measurement [17]. | Re-clean the cuvette, perform a new blank, and re-read the sample. | |
| Inconsistent Replicate Readings | Cuvette orientation is not consistent [17]. | Always insert the cuvette with the same orientation facing the light path. |
| Sample is degrading (light-sensitive or evaporating) [17]. | Take readings quickly after prep; keep cuvette covered to prevent evaporation. |
Experimental Protocols for Drift Mitigation
Protocol 1: Establishing an Environmental Monitoring and Control Procedure
Objective: To systematically monitor and control the laboratory environment to minimize its contribution to instrumental drift.
Materials:
- Field Spectrometer
- Calibrated, data-logging thermometer/hygrometer
- Vibration isolation platform (optional but recommended)
- Laboratory notebook
Methodology:
- Baseline Data Collection: Place the thermometer/hygrometer next to the spectrometer. Over a 48-hour period without instrument operation, record the temperature and humidity at regular intervals (e.g., every 30 minutes) to establish ambient environmental fluctuations.
- Identify Stable Locations: Analyze the data to identify locations in the lab with the smallest temperature fluctuations and lowest levels of vibration. Avoid areas near air conditioning vents, heaters, doors, or high-traffic pathways [17].
- Instrument Placement: Place the spectrometer on a stable, level bench in the identified optimal location. For areas with inherent vibration, use a vibration isolation platform [52].
- Pre-Operational Equilibration: On days of use, turn on the spectrometer and allow it to warm up for 30 minutes. Meanwhile, record the current temperature and humidity.
- In-Operation Monitoring: During critical measurements, continue to monitor environmental conditions. If significant deviations from the stable baseline are observed, note them in the experiment log as they may be a source of drift.
Protocol 2: Systematic Sample Preparation to Minimize Artifacts
Objective: To ensure sample preparation techniques do not introduce noise or drift into spectral measurements.
Materials:
- High-purity solvents and reagents
- Matched quartz or glass cuvettes
- Lint-free gloves and lint-free wipes
- Pipettes and volumetric flasks
Methodology:
- Cuvette Handling: Always handle cuvettes by the frosted or ribbed sides to prevent fingerprints on the optical surfaces [17].
- Cleaning: Before use, clean cuvettes with a series of rinses using high-purity solvent. Dry the outside with a lint-free wipe. Visually inspect to ensure they are clean, scratch-free, and without residue.
- Blanking: Fill the cuvette with the pure solvent or buffer used for sample dissolution. Use this same cuvette for the blank measurement to establish the baseline [17].
- Sample Preparation: Prepare your sample in the appropriate solvent. Ensure it is fully dissolved and homogeneous.
- Loading: Pour the prepared sample into a clean, dry cuvette (ideally the same one used for blanking). Check for and gently tap the cuvette to dislodge any air bubbles, as they scatter light and cause errors [17].
- Measurement: Place the cuvette in the holder in a consistent orientation every time. Close the sample compartment lid and begin measurement.
Research Reagent Solutions for Drift Management
The following table details essential materials and reagents used to maintain accuracy and combat drift.
| Item | Function / Rationale |
|---|---|
| NIST-Traceable Calibration Standards (e.g., Holmium Oxide filter, neutral density filters) | Certified reference materials provide an absolute reference to verify the photometric and wavelength accuracy of the spectrometer, detecting inherent instrument drift [53]. |
| High-Purity, Spectral Grade Solvents | Solvents with low UV absorbance and minimal impurities prevent introducing baseline noise and ghost peaks that can mask true sample signals or be mistaken for drift [54] [55]. |
| Matched Quartz Cuvettes | For UV measurements, quartz is mandatory. Using a matched pair ensures the blank and sample are measured in optically identical paths, preventing artifacts like negative absorbance [17]. |
| Stable Reference Probe | A sensor designed to be isolated from the primary measurement variable (e.g., pressure) but exposed to the same environmental conditions (e.g., temperature). Its highly correlated output can be used to mathematically correct for drift in the primary sensor [2]. |
| Lint-Free Wipes & Powder-Free Gloves | Prevents contamination of optical surfaces (cuvettes, reference tiles) with fibers, oils, or particulates that scatter light and cause inaccurate readings [53]. |
Drift Investigation Workflow
The diagram below outlines a systematic logical workflow for investigating the source of instrumental drift.
Developing Robust Correction Functions for Variable Field Conditions
FAQs: Core Concepts and Challenges
Q1: What is calibration transfer and why is it critical for field spectroscopy?
Calibration transfer is the process of applying a predictive model developed on one spectrometer (the "master") to data collected from another spectrometer (the "slave") or under different conditions, without a significant loss in performance [14]. This is crucial because models trained on one instrument often fail when applied to others due to hardware-induced spectral variations, which can stem from differences in wavelength alignment, spectral resolution, and detector noise [14]. Successful calibration transfer is essential for ensuring reliable and consistent measurements across different field instruments and over time.
Q2: What are the primary sources of spectral variability in field conditions?
Spectral data can vary significantly due to a range of factors, which can be broadly categorized as follows [14]:
- Instrument-Related Variability: This includes wavelength alignment errors, spectral resolution and bandwidth differences, and detector noise variability. Even nominally identical instruments can produce different spectral data due to these hardware factors [14].
- Environmental Variability: In field spectroscopy, changing illumination conditions are a major challenge. The proportion of diffuse radiance and the direction of direct sunlight can alter the apparent reflectance of a target [56]. Temperature and humidity can also affect instrument components and sample properties.
- Long-Term Temporal Drift: Instrumental response can change over extended periods (e.g., days or months) due to factors like instrument power cycling, component aging (e.g., lamp intensity), column replacement in chromatographs, or contamination of optical parts [7].
Q3: My spectrometer's readings are unstable and drifting. What are the most common causes?
Unstable or drifting readings can often be traced to a few common issues. The table below summarizes these causes and their solutions [17].
| Possible Cause | Recommended Solution |
|---|---|
| Insufficient lamp warm-up | Allow the instrument to warm up for at least 15-30 minutes before use to let the light source stabilize [17]. |
| Changing illumination (for field reflectometry) | Use a dual spectrometer system that simultaneously measures the target and a white reference to correct for fluctuating sunlight [56]. |
| Air bubbles in the sample | Gently tap the cuvette to dislodge bubbles before measurement [17]. |
| Environmental factors | Place the spectrometer on a stable, level surface away from vibrations, drafts, or large temperature fluctuations [17]. |
| Aging or failing light source | Check the lamp's usage hours and replace it if it is nearing the end of its life [17]. |
| Dirty optical windows or fibers | Clean the windows located in front of the fiber optic and in the direct light pipe according to the manufacturer's instructions [3]. |
Troubleshooting Guides
Correcting for Long-Term Instrumental Drift
Long-term drift is a critical challenge for tracking processes or product stability over weeks or months. The following protocol, adapted from GC-MS research, provides a robust methodology for correction using Quality Control (QC) samples [7].
Experimental Protocol: QC-Based Drift Correction
- Objective: To correct for signal drift in quantitative measurements over an extended period (e.g., 155 days).
- Materials:
- Pooled QC sample that contains all, or a representative subset of, the chemicals present in your test samples.
- Virtual QC Sample: A meta-reference created by combining chromatographic peaks from all QC measurements, verified by retention time and mass spectrum [7].
- Methodology:
- Data Collection: Conduct repeated measurements of your test samples and the pooled QC sample over the entire study period. Record the batch number (tracking instrument on/off cycles) and injection order number for each measurement [7].
- Calculate Correction Factors: For each component (e.g., chemical peak)
kin the QC, calculate its correction factoryfor measurementias:y_i,k = X_i,k / X_T,k, whereX_i,kis the peak area in measurementiandX_T,kis the median peak area across all QC measurements [7]. - Model the Drift: Model the correction factor
y_kas a function of batch numberpand injection ordert:y_k = f_k(p, t). Three algorithms were evaluated for this purpose [7]:- Random Forest (RF): Provided the most stable and reliable correction for long-term, highly variable data.
- Support Vector Regression (SVR): Tended to over-fit and over-correct data with large variations.
- Spline Interpolation (SC): Exhibited the lowest stability for the dataset in the study.
- Apply the Correction: For a test sample, input its batch number and injection order into the correction function
f_kto get the predicted coefficienty. The corrected peak area is then calculated as:x'_k = x_k / y[7].
The workflow for implementing this correction strategy is summarized in the following diagram:
Addressing Flat Field and Stray Light Artifacts
Non-uniform detector sensitivity and stray light can severely distort spectral data, masking weaker signals. Flat field correction is a key method to address this.
Experimental Protocol: Flat Field Correction for a Spatial Heterodyne Spectrometer
This procedure details the steps for a specific spectrometer type, but the principle is widely applicable to array-based sensors [57].
- Objective: To remove pixel-to-pixel variations in instrument/detector sensitivity.
- Materials:
- A calibrated, broad-band white light source.
- Methodology:
- Collect Raw Image Frames (
I_R): Collect and average multiple raw image frames of your sample [57]. - Collect Dark Frame (
D): Cover the spectrometer's input and collect multiple images to capture the detector's noise profile. Average these frames [57]. - Collect Flat Frame (
F): Image the broad-band white light source. For interferometric systems, this may need to be done separately for each grating to remove interference fringes. Average these frames [57]. - Apply Correction: Use the following equation to generate the corrected image
I_C:I_C = (I_R - D) / (F - D)[57]. This process removes the instrument's response profile, revealing a cleaner spectrum.
- Collect Raw Image Frames (
Effect of Flat Field Correction on Spectral Data The application of flat field correction can significantly improve data quality, as demonstrated in a study on a potassium nitrate sample [57].
| Metric | Uncorrected Data | Flat Field Corrected Data |
|---|---|---|
| Baseline Noise | High, obscuring smaller peaks | Majorly reduced throughout the spectrum |
| Signal-to-Noise Ratio (SNR) | Baseline for comparison | Improved by a factor of 1.6 |
| Detection of Weak Peaks | A cluster of peaks at ~530 pixels was not detectable | Peaks successfully detected above the noise floor |
The Scientist's Toolkit: Research Reagent Solutions
The following table lists key materials and computational tools referenced in the protocols for developing robust correction functions.
| Reagent / Solution | Function in Experiment |
|---|---|
| Pooled Quality Control (QC) Sample | Serves as a standardized reference for tracking and correcting instrumental drift over time; should be representative of the sample matrix [7]. |
| Spectralon/Zenith Polymer Target | A near-perfect diffuse reflector with a flat spectral response, used as a white reference to convert field measurements to absolute reflectance [56]. |
| Virtual QC Sample | A computational reference created from the median of all QC runs, providing a stable meta-reference for normalization that accounts for overall study variance [7]. |
| Random Forest Algorithm | A machine learning algorithm used to build a stable and reliable model for correcting long-term, highly variable instrumental drift [7]. |
| Flat Field Image | An image of a uniform light source used to characterize and correct for pixel-to-pixel variations in detector sensitivity [57]. |
Creating a Proactive Maintenance and Quality Assurance Protocol
For researchers relying on field spectrometers, calibration drift is not merely an inconvenience; it is a significant source of data error that can compromise research validity, particularly in long-term environmental monitoring and precise analytical studies in drug development. A proactive maintenance and quality assurance protocol is essential to identify and correct the root causes of drift before they manifest as inaccurate results. This approach moves beyond reactive fixes to embrace a strategy of prevention, leveraging scheduled checks, condition monitoring, and data-driven interventions to ensure instrument reliability and data integrity over time [58] [59]. This guide provides a foundational framework, including troubleshooting FAQs and experimental protocols, to help scientists maintain the accuracy of their spectroscopic measurements.
Foundational Maintenance Strategies
A proactive protocol is built on a combination of scheduled and condition-based activities designed to prevent failures.
Core Strategy Types
| Strategy Type | Brief Description | Example for a Field Spectrometer |
|---|---|---|
| Preventive Maintenance | Maintenance performed at scheduled, predetermined intervals based on calendar time or instrument usage [58] [60]. | Quarterly cleaning of optical windows and annual replacement of the light source, regardless of current performance. |
| Condition-Based Maintenance | Maintenance triggered by based on the monitored condition of the equipment, indicating a change in its state [58] [59]. | Monitoring light source intensity or signal-to-noise ratio; performing maintenance when values deviate from a defined baseline. |
| Predictive Maintenance | An advanced form of condition-based maintenance that uses data analysis and predictive models to forecast potential failures [60] [59]. | Using historical drift data and machine learning to predict the remaining useful life of a critical sensor component. |
Proactive Maintenance Workflow
The following workflow outlines a continuous cycle for maintaining spectrometer performance and preventing calibration drift.
Diagram: Proactive Maintenance Workflow for Spectrometers
Troubleshooting Guide & FAQs
This section addresses common spectrometer issues that can lead to calibration drift and inaccurate data.
Frequently Asked Questions
Q1: My spectrometer's readings are unstable and drift over time during a measurement session. What could be the cause?
- A: Drifting readings can stem from several issues [17] [61]:
- Insufficient Warm-Up: Ensure the instrument's lamp has been allowed to stabilize for at least 15-30 minutes after being turned on.
- Environmental Factors: Place the spectrometer on a stable, level surface away from vibrations, drafts, and significant temperature fluctuations.
- Contaminated Gas/Purge: For instruments requiring a purged environment, contaminated or humid purge gas can cause drift in certain wavelengths. Ensure argon or nitrogen supplies are pure and gas lines are clean [3].
- Aging Light Source: Lamps lose intensity over time. Check the usage hours in the instrument's software and replace the lamp if it is near or beyond its rated lifespan.
Q2: The instrument fails to zero or blank properly. How should I troubleshoot this?
- A: Blanking errors are often related to the sample compartment or the blank itself [17]:
- Compartment Lid: Verify that the sample compartment lid is fully closed, as external light leakage will prevent proper zeroing.
- Optical Windows: Check that the optical windows in the sample path are clean. Contamination can scatter light and cause poor blanking.
- Cuvette and Blank: Use the exact same, clean cuvette for both the blank and sample measurements. Ensure the blank solution is the correct one for your experiment (e.g., the same buffer your sample is in, not just water).
Q3: I observe inconsistent results between replicate sample measurements. What is the most likely source of this error?
- A: Inconsistency often points to sample handling or preparation [17]:
- Cuvette Orientation: Always place the cuvette in the holder in the same orientation. Slight optical variations between the cuvette's faces can cause signal variance.
- Air Bubbles: Gently tap the cuvette after filling to dislodge any air bubbles, which scatter light and create erratic readings.
- Sample Degradation: If the sample is light-sensitive or reacts over time, measurements may change between replicates. Minimize the time between readings.
Q4: How does water vapor specifically affect spectroscopic measurements, and how can it be corrected?
- A: Water vapor causes spectral interference, particularly in infrared and cavity ring-down spectroscopy, leading to substantial biases in measurements like δ¹³CH₄ [15]. It can broaden absorption lines and contribute to the signal at key wavelengths. Correction requires:
- Drying the Sample: Using a Nafion dryer to remove humidity from the air sample before analysis.
- Empirical Correction: Establishing a humidity correction function in the laboratory. Research shows that applying a linear correction for δ¹³CH₄ and quadratic corrections for ¹²CH₄ and ¹³CH₄ can effectively remove humidity-induced biases [15].
Experimental Protocol: Correcting for Calibration Drift
This protocol provides a methodology for quantifying and correcting for inherent calibration drift in field spectrometers, inspired by principles of in-situ correction used in deep-sea sensors [2].
Workflow for Drift Assessment and Correction
The experiment follows a systematic process from setup to data correction, as visualized below.
Diagram: Experimental Workflow for Drift Correction
Detailed Methodology
Objective: To characterize the temporal drift of a field spectrometer and develop a mathematical function to correct field data.
Materials & Reagents:
- Field Spectrometer
- Set of Certified Calibration Standards (e.g., neutral density filters, rare earth oxide solutions)
- Environmental Chamber (capable of controlling temperature and humidity)
- Data Logging Software
- Statistical Analysis Software (e.g., R, Python)
Procedure:
Initial Calibration:
- Allow the spectrometer to warm up for the manufacturer-specified time (typically 30+ minutes) in a controlled environment [17].
- Measure the certified calibration standards in a randomized order. Replicate each measurement three times.
- Construct a calibration curve (e.g., Signal vs. Known Value) for each wavelength or channel of interest. Record the slope, intercept, and R² value.
Drift Simulation and Monitoring:
- Place the spectrometer in an environmental chamber set to simulate field conditions (e.g., 25°C, 50% RH).
- At regular intervals (e.g., daily for the first week, then weekly), repeat the measurement of the calibration standards without adjusting the instrument's calibration.
- Log all measurements alongside timestamp and environmental data (temperature, humidity).
Data Analysis:
- For each time interval, calculate the apparent value of each standard using the initial calibration curve.
- The difference between the apparent value and the known standard value represents the drift.
- Model the drift over time. Drift is often linear or monotonic in the short term but may require more complex models (e.g., ARIMA) for long-term forecasting [2]. A simple linear model is:
Drift(t) = a * t + b, wheretis time.
Correction Function:
- The inverse of the drift model becomes the correction function. For a linear drift, the correction for a field measurement
M_fieldtaken at timetis:M_corrected = M_field - (a * t + b).
- The inverse of the drift model becomes the correction function. For a linear drift, the correction for a field measurement
Key Quantitative Data from Drift Studies
Table: Summary of Drift Correction Insights from Research
| Factor | Impact on Drift | Correction Method & Efficacy | Source |
|---|---|---|---|
| Water Vapor (1.5% - 4.0%) | Induces substantial bias in δ¹³CH₄ measurements. | Applying a linear water vapor correction function successfully removed biases. | [15] |
| Long-Term Deployment | Optical pressure sensors experienced drift in deep-sea environments. | An in-situ correction method using a reference probe maintained stability within ± 0.033% F.S. | [2] |
| Calibration Interval | Drift accumulates over time, increasing measurement uncertainty. | Analysis recommended a calibration interval of no more than six months for long-term accuracy. | [2] |
The Scientist's Toolkit: Essential Reagents & Materials
Table: Key Materials for Spectrometer Maintenance and Drift Studies
| Item | Function / Purpose |
|---|---|
| Certified Reference Standards | Provide a known, stable signal to validate instrument calibration and quantify drift over time. |
| Nafion Dryer | Removes water vapor from air samples to eliminate spectral interference and humidity-induced bias in gas measurements [15]. |
| Stable Light Source | An external, intensity-stable lamp (e.g., calibrated deuterium lamp) used for independent verification of the instrument's photometric stability. |
| Lint-Free Wipes | For cleaning optical windows and cuvettes without introducing scratches or fibers that can scatter light. |
| Quartz Cuvettes | Required for UV range measurements (<340 nm); glass and plastic cuvettes absorb UV light [17]. |
| Optically Matched Cuvette Set | Ensures that the pathlength and optical properties are identical between the cuvette used for blanking and the one used for samples, reducing error. |
| Desiccant | Used in instrument storage compartments to control internal humidity and protect sensitive optical components. |
Validating Correction Methods: Performance Metrics and Case Studies
Frequently Asked Questions (FAQs)
Q1: My field spectrometer's readings are consistently offset from the reference values. Which metric should I use to diagnose this? A1: The Bias metric is the most appropriate for identifying a consistent offset or systematic error. Bias measures the average difference between your predicted values and the known reference values, indicating whether your instrument is consistently reading high or low [62] [63]. A statistically significant bias often necessitates a bias correction in your calibration model [63].
Q2: How can I tell if my calibration model is accurate for predicting new, unknown samples, not just the ones it was built with? A2: To evaluate predictive accuracy, you should calculate the Root Mean Square Error of Prediction (RMSEP) [62] [64]. Unlike the Standard Error of Calibration (SEC), which tests the model on the same samples used to create it, RMSEP is calculated using a separate, independent set of validation samples. This mimics real-world application and is considered the best measure of calibration quality for predicting unknowns [62].
Q3: My sensor data shows large, occasional spikes. How will this affect my error metrics? A3: Large spikes, or outliers, have a significant impact on Mean Squared Error (MSE) and Root Mean Squared Error (RMSE) because these metrics square the errors before averaging [65]. This squaring effect emphasizes larger errors. If your data is prone to outliers and you want a more robust model, optimizing for RMSE might be preferable. If your primary goal is the highest precision and you want to heavily penalize large errors, MSE is the more suitable metric [65].
Q4: How often should I recalibrate my field spectrometer to correct for long-term drift? A4: Recalibration frequency depends on the instrument and environmental stresses. A 30-month field study on NDIR CO₂ sensors found that drifts can produce biases of up to 27.9 ppm over two years [16]. The study recommends a calibration frequency preferably within 3 months and not exceeding 6 months to maintain accuracy. For optimal results, perform calibration during different seasonal conditions, such as both winter and summer [16].
Troubleshooting Guides
Problem: Declining Predictive Accuracy Due to Calibration Drift
Symptoms:
- Gradual increase in the Root Mean Square Error of Prediction (RMSEP) or Bias over time when testing with validation samples [16] [62].
- The calibration model performs well on old data but poorly on newly collected samples.
Investigation and Resolution Steps:
- Verify Drift: Co-locate your field spectrometer with a high-precision reference instrument or known standard for a short period. Calculate the Bias and RMSE between your sensor data and the reference data. A significant bias or increase in RMSE confirms drift [16] [66].
- Identify Drift Type:
- Apply Correction:
- For long-term linear drift, linear interpolation between periodic calibrations can be an effective correction method, significantly reducing long-term RMSE [16].
- For environmental influences, develop a multivariate regression model that incorporates temperature, pressure, and humidity to correct the raw sensor data [16] [66].
Symptoms:
- High Root Mean Square Error (RMSE) or Standard Error of Calibration (SEC) values, indicating poor model fit and large average errors [16] [62].
Investigation and Resolution Steps:
- Check Environmental Sensitivity: Raw data from sensors like NDIR spectrometers are often highly sensitive to environmental factors. A controlled laboratory test can quantify the impact of temperature, pressure, and humidity on your readings [16] [66].
- Apply Environmental Correction: Implement a multivariable linear regression calibration that uses measured temperature, pressure, and humidity as independent variables to correct the CO₂ mole fraction (or other target analyte) reading. This can reduce RMSE dramatically [66].
- Validate the Correction: After applying the environmental correction, re-calculate the RMSE and Bias by comparing your corrected sensor data to reference data. A successful correction will show these metrics falling within acceptable limits [16] [66].
The following tables summarize key performance metrics from recent studies on sensor calibration and drift correction.
Table 1: Performance of Sensor Calibration Methods in Field and Laboratory Settings
| Study Focus | Sensor / Instrument | Calibration Method | Key Performance Metric | Result Before Correction | Result After Correction |
|---|---|---|---|---|---|
| CO₂ Sensor Field Performance [16] | SENSE-IAP (NDIR) | Environmental Correction | Root Mean Square Error (RMSE) | 5.9 ± 1.2 ppm | 1.6 ± 0.5 ppm |
| CO₂ Sensor Long-Term Drift [16] | SENSE-IAP (NDIR) | Linear Interpolation | RMSE over 30 months | ~27.9 ppm (bias) | 2.4 ± 0.2 ppm |
| NDIR CO₂ Sensor Calibration [66] | Vaisala GMP343 | Multivariable Linear Regression (Lab) | RMSE / Bias | 5.218 ppm / N/R | 2.1 ppm / 0.003 ppm |
| NDIR CO₂ Sensor Calibration [66] | Vaisala GMP343 | Multivariable Linear Regression (Field) | RMSE / Bias | 8.315 ppm / 39.170 ppm | 2.154 ppm / 0.018 ppm |
| LIBS Wavelength Calibration [26] | MarSCoDe LIBS | Matching Global Iterative Registration | Internal Accord Accuracy (RMSE in pixels) | N/R | 0.292, 0.223, 0.247 pixels |
Table 2: Key Error Metrics and Their Definitions in Calibration Science
| Metric Name | Acronym | Formula | Interpretation in Calibration Context |
|---|---|---|---|
| Root Mean Square Error [65] [64] | RMSE | RMSE = √[ ∑(y_i - ŷ_i)² / n ] |
The standard deviation of the prediction errors. Indicates how concentrated the data is around the line of best fit. |
| Root Mean Square Error of Prediction [62] [64] | RMSEP | RMSEP = √[ ∑(y_i - ŷ_i)² / p ] |
The standard deviation of prediction errors for an independent validation set. Best measure of real-world model performance. |
| Standard Error of Calibration [62] | SEC | SEC = √[ ∑(P_i - K_i)² / n ] |
The standard deviation of (predicted - known) values for the calibration sample set. Measures how well the model fits its own data. |
| Bias [62] [63] | Bias | Bias = ∑(P_i - K_i) / n |
The average difference between predicted (Pi) and known (Ki) values. Measures systematic error or consistent offset. |
| Mean Squared Error [65] | MSE | MSE = ∑(y_i - ŷ_i)² / n |
The average of the squares of the errors. Strongly penalizes large errors due to the squaring function. |
Experimental Protocols
Detailed Methodology: Multivariable Linear Regression for Environmental Correction
This protocol is adapted from studies calibrating low-cost NDIR CO₂ sensors and is applicable to field spectrometers affected by environmental variables [16] [66].
1. Goal: To develop a calibration model that corrects for the effects of temperature (T), pressure (P), and relative humidity (RH) on spectrometer readings.
2. Materials and Setup:
- Unit Under Test (UUT): The field spectrometer to be calibrated.
- Reference Instrument: A high-precision analyzer (e.g., Picarro cavity ring-down spectrometer) traceable to a primary standard [16] [66].
- Environmental Chamber: A chamber capable of controlling and varying T, P, and RH [66].
- Data Logging System: To synchronously record data from the UUT, reference instrument, and environmental sensors.
3. Procedure:
- Co-located Measurement: Place the UUT and the reference instrument in the environmental chamber. Ensure they are measuring the same air sample or standard.
- Environmental Stressing: Over a set period, systematically vary the environmental conditions within the chamber to cover the expected operational range of the UUT (e.g., temperature from -10°C to 24°C, pressure from 978 hPa to 1032 hPa, RH from 56% to 89%) [66].
- Data Collection: Continuously record the synchronized outputs of the UUT, the reference instrument, and the environmental sensors (T, P, RH).
- Model Development: Perform a multivariate linear regression analysis where the reference instrument's reading is the dependent variable, and the UUT's raw reading plus the environmental variables are the independent variables [16] [66]. The model form is typically:
[Reference Value] = β₀ + β₁*[Raw UUT] + β₂*[T] + β₃*[P] + β₄*[RH].
4. Validation:
- Reserve a portion of the collected data not used in model development for validation.
- Apply the derived model to the raw UUT validation data.
- Calculate RMSEP and Bias between the model-corrected UUT data and the reference data to quantify the improvement in accuracy [62].
Detailed Methodology: Assessing and Correcting Long-Term Drift
This protocol outlines a strategy for monitoring and correcting gradual sensor drift over time, as demonstrated in long-term field evaluations [16].
1. Goal: To quantify long-term drift and establish a recalibration schedule.
2. Materials and Setup:
- Deployed field spectrometers.
- A single, stable, and traceable reference standard or a high-precision reference instrument for periodic co-located measurements.
3. Procedure:
- Establish Baseline: Perform an initial, high-quality calibration of all field units against the reference standard.
- Periodic Co-location: At regular intervals (e.g., every 1-3 months), bring a field unit to a central location to be co-located with the reference standard for a short period (e.g., 10 days) [16] [66].
- Drift Calculation: For each co-location period, calculate the Bias and RMSE between the field unit and the reference.
- Correction Model: Model the drift over time. A simple and effective method is linear interpolation of the bias between calibration points [16]. For a sensor calibrated at time T1 and T2, the drift correction at time T (where T1 < T < T2) is applied based on the interpolated bias.
4. Determining Recalibration Frequency:
- The recalibration frequency is determined by the rate at which the sensor's error (e.g., RMSE or Bias) exceeds the required accuracy threshold for your application. Research suggests that for certain CO₂ sensors, this should not exceed 6 months to ensure accuracy within 5 ppm [16].
Workflow Visualization
Troubleshooting Workflow for Calibration Performance
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Spectrometer Calibration and Validation
| Item | Function in Research | Example from Literature |
|---|---|---|
| High-Precision Reference Analyzer | Serves as the "ground truth" to calibrate against and validate the performance of field-deployed sensors. | Picarro cavity ring-down spectroscopy (CRDS) analyzers are used as reference instruments for CO₂ monitoring networks [16] [66]. |
| Environmental Chamber | Allows for controlled testing and characterization of a sensor's sensitivity to temperature, pressure, and humidity in a laboratory setting. | Used to apply a multivariable linear regression calibration to NDIR CO₂ sensors by varying T, P, and RH [66]. |
| Stable Isotope-Labeled Internal Standards | Added to samples in mass spectrometry to compensate for matrix effects and inefficiencies in sample preparation, improving accuracy and precision [67]. | Used in LC-MS/MS clinical mass spectrometry procedures to mitigate the impact of matrix ion suppression or enhancement [67]. |
| Matrix-Matched Calibrators | Calibration standards prepared in a matrix that closely resembles the patient or sample matrix, reducing bias caused by matrix differences [67]. | Recommended for clinical mass spectrometry to avoid biased measurements when quantifying endogenous analytes [67]. |
| Wavelength & Photometric Standards | Stable reference materials with known photometric values or emission lines used to calibrate the wavelength and photometric response (absorbance/reflectance) axes of a spectrometer [68] [26]. | Fluorilon R99 is used to assess photometric accuracy of NIR spectrophotometers [68]. Titanium-alloy samples are used for on-board wavelength calibration of the MarSCoDe LIBS instrument on Mars [26]. |
| Validation Sample Set | An independent set of samples with known analyte concentrations that were not used in the calibration process. Used to calculate RMSEP and test real-world predictive ability [62]. | A set of validation samples (minimum of 3) is used to calculate the Standard Error of Prediction (SEP) for an IPA-in-water calibration model [62]. |
For researchers and scientists working with field spectrometers and analytical instruments, calibration drift is a pervasive and critical challenge. It refers to the gradual, often unpredictable change in an instrument's response over time, leading to systematic errors and compromising data integrity [69]. This phenomenon can be caused by sensor aging, environmental fluctuations, material degradation, and changes in sample matrices [70]. For professionals in drug development and analytical research, uncorrected drift can invalidate long-term studies, lead to faulty conclusions, and impact product quality.
To combat this, advanced algorithmic correction methods have been developed. This guide provides a comparative analysis of three prominent algorithms—Spline Interpolation (SC), Support Vector Regression (SVR), and Random Forest (RF)—to help you select and implement the most effective strategy for your experimental conditions.
Troubleshooting Guides & FAQs
Frequently Asked Questions
Q1: My spectrometer's readings for the same reference material are changing over several months. What is this phenomenon? This is典型的校准漂移。类似于 [20]中描述的情况,即仪器即使测量同一种物质,也会产生不准确的结果范围。漂移是由传感器老化、环境条件(如温度、湿度)变化以及电子元件不稳定等因素引起的系统性偏差 [69] [70]。
Q2: What is the fundamental difference between a 'correction' algorithm and simply recalibrating my instrument? Recalibration typically involves a full, often manual, procedure using a set of reference materials to reset the instrument's baseline, which can be labor-intensive and interrupt operation [70]. A correction algorithm, in contrast, uses a mathematical model to digitally adjust the raw output data from your instrument. This leverages historical data from Quality Control (QC) samples to compensate for drift without always requiring physical intervention [7].
Q3: When should I consider using a machine learning-based algorithm like Random Forest over a simpler method? You should consider advanced methods like Random Forest when dealing with complex, non-linear drift patterns, when multiple environmental factors influence the drift simultaneously, or when traditional methods fail to provide stable corrections across the entire measurement range [36] [7]. Studies have shown that machine learning models can maintain calibration better over time compared to regression models [36].
Troubleshooting Common Algorithm Implementation Issues
Problem: After applying my correction algorithm, the results for high-concentration samples are still inaccurate.
- Potential Cause: The algorithm may not be capturing non-linear effects that are more pronounced at extreme concentrations. This was observed in delta-based calibrations for methane, where performance was compromised by non-linear spectral effects at high concentrations [15].
- Solution: Ensure your training dataset, especially your QC samples, adequately covers the entire concentration range you expect to measure. Consider switching to an algorithm like Random Forest, which is better at handling non-linear relationships [7].
Problem: My correction model works well in the lab but fails when deployed in the field.
- Potential Cause: The model has overfitted to the laboratory conditions and cannot generalize to the new, variable environment (e.g., different humidity, temperature). This highlights the difference between dried and humid air measurements, as significant biases can occur in humid conditions without a robust strategy [15].
- Solution: Incorporate data from field conditions into your training set. Techniques like Domain-Adversarial Learning are specifically designed to make models robust to such shifts in data distribution [70].
Problem: The correction algorithm introduces more noise and variability into the data.
- Potential Cause: This is a classic sign of over-fitting, where the model learns the noise in the training data rather than the underlying drift signal. The study on GC-MS data noted that SVR, in particular, tends to over-fit and over-correct highly variable data [7].
- Solution: Simplify your model or use regularization techniques. You might also increase the number of QC samples used for training to provide a more robust signal. Random Forest was found to provide the most stable correction in such scenarios [7].
Experimental Protocols for Algorithm Evaluation
Implementing a drift correction strategy requires a structured experimental approach. The following protocol, inspired by long-term GC-MS studies [7], provides a robust framework.
Protocol: Establishing a Drift Correction Model Using QC Samples
Objective: To collect data for building and validating SC, SVR, and Random Forest drift correction models for an analytical instrument over an extended period.
Essential Materials and Reagents: Table: Key Research Reagent Solutions
| Item | Function in Experiment |
|---|---|
| Certified Reference Materials (CRMs) | To perform initial instrument calibration and establish a ground-truth baseline. |
| Pooled Quality Control (QC) Sample | A homogeneous sample analyzed at regular intervals to track instrumental drift over time [7]. |
| Drift Monitors | Specialized, stable materials used to assess the stability of instruments like XRF spectrometers and support drift correction [20] [46]. |
| Test Samples | A set of samples with known properties, used to validate the performance of the correction models. |
Methodology:
- Initial Calibration: Calibrate the instrument using CRMs to ensure it is in an optimal baseline state.
- QC Sample Preparation: Create a large, homogeneous batch of a pooled QC sample. This sample should be representative of the analytes you typically measure.
- Long-Term Data Acquisition:
- Over an extended period (e.g., 155 days as in [7]), periodically analyze the QC sample. The interval (e.g., daily, weekly) should be frequent enough to capture drift dynamics.
- In the same sequence, analyze your validation test samples.
- Record all peak areas or relevant response data, along with two critical metadata points:
- Data Processing:
- For each analyte in the QC sample, calculate a "true value" (e.g., the median response,
X_T,k) from all the QC runs. - For each QC measurement
iof analytek, compute the correction factor:y_i,k = X_i,k / X_T,k[7].
- For each analyte in the QC sample, calculate a "true value" (e.g., the median response,
- Model Training: Use the dataset of
{p, t, y_i,k}to train the three correction models (SC, SVR, RF). The models learn the functiony_k = f_k(p, t).
The workflow for this protocol is summarized in the following diagram:
Quantitative Algorithm Comparison
The following table synthesizes quantitative and performance data from a direct comparative study of SC, SVR, and Random Forest for correcting GC-MS drift over 155 days [7]. This provides a clear, evidence-based summary for decision-making.
Table: Performance Comparison of SC, SVR, and Random Forest Correction Algorithms
| Algorithm | Core Principle | Stability & Reliability | Handling of Non-Linearity | Risk of Over-fitting | Best-Suited Application Context |
|---|---|---|---|---|---|
| Spline Interpolation (SC) | Uses segmented polynomials (e.g., Gaussian) to interpolate between data points [7]. | Lowest stability; fluctuations with sparse QC data [7]. | Limited to the interpolation function used. | Low | Short-term studies with very high data density and simple, monotonic drift. |
| Support Vector Regression (SVR) | Finds an optimal hyperplane to perform regression for continuous function prediction [7]. | Less stable than RF; performance degrades with high variability [7]. | Good, but can be compromised by over-fitting. | High - tends to over-fit and over-correct highly variable data [7]. | Scenarios with a clear, smooth drift function and ample training data to mitigate over-fitting. |
| Random Forest (RF) | Supervised machine learning using an ensemble of decision trees [36] [71] [7]. | Most stable and reliable for long-term, highly variable data [7]. | Excellent - naturally captures complex, non-linear relationships [71] [7]. | Low - robust due to ensemble approach. | Complex, long-term studies with non-linear drift, multiple influencing factors, and a need for robust performance [36] [7] [70]. |
Key Recommendations for Scientists
Based on the comparative analysis, here are actionable recommendations for different research scenarios:
For Most Long-Term Studies: Prioritize Random Forest. Its proven stability and ability to handle non-linear drift without over-fitting make it the most reliable choice for extended experiments, such as those in drug stability testing or long-term environmental monitoring [7] [70].
For Short-Term or Linearly-Drifting Systems: If your preliminary data shows a simple, linear drift and your QC measurements are frequent, Spline Interpolation can be a computationally simple and effective solution.
When Data is Abundant and Well-Understood: Support Vector Regression can be a powerful tool, but it requires careful tuning and validation to prevent over-fitting. Use it when you have a deep understanding of the drift characteristics in your system.
Universal Requirement: Robust QC. Regardless of the algorithm you choose, the foundation of successful drift correction is a rigorous QC protocol. This includes using a consistent, representative pooled QC sample and tracking critical metadata like batch and injection order [7].
Troubleshooting Guide: Identifying and Correcting Spectrometer Drift
This guide helps users diagnose and address common spectrometer drift issues encountered during long-term field deployment.
Q1: My spectrometer's results for the same sample are becoming inconsistent over time. What is happening? Symptom: Increasingly varied results for the same certified reference material (CRM) or control sample. Potential Cause: Instrument drift, where the spectrometer's calibration shifts from its original baseline due to environmental factors or component aging [20]. Troubleshooting Steps:
- Verify with Control Samples: Measure a known control sample. If results fall outside established tolerance limits, drift is likely occurring [72].
- Inspect the Vacuum Pump (for OES): A malfunctioning vacuum pump can cause low-intensity readings for elements like Carbon, Phosphorus, and Sulfur. Check for pump issues like overheating, unusual noises, or oil leaks [3].
- Check for Contaminated Argon or Samples: Contaminated argon can cause a white, milky burn appearance and unstable results. Ensure samples are not contaminated by oils from skin or quenching processes [3].
- Clean Optical Windows: Dirty windows on the fiber optic or light pipe can cause analytical drift and poor results. Clean these windows as part of regular maintenance [3].
Q2: How can I confirm if my instrument's spectral calibration has drifted? Symptom: Identifiable emission peaks or absorption features appear at incorrect wavelengths in your spectrum. Diagnostic Method: Use an on-board calibration system. For example, the NEON Imaging Spectrometer (NIS) uses a mercury lamp to verify that its spectral calibration has not shifted [73]. Corrective Action: Perform a wavelength recalibration using a certified emission source (e.g., Hg-Ar lamp) with known peak positions [4].
Q3: What are the primary environmental factors causing drift in field spectrometers? Field instruments are particularly susceptible to temperature fluctuations [74]. Temperature changes affect electronic components like scintillation crystals and photomultiplier tubes, leading to significant spectrum drift [74]. Mitigation Strategy:
- Gain Adjustment: Implement a correction method that combines real-time gain adjustment with post-processing algorithms to counteract temperature-induced drift [74].
- Stabilize Temperature: If possible, use instruments with temperature-stabilized detectors or housings.
Frequently Asked Questions (FAQs)
Q: What is the difference between a Certified Reference Material (CRM) and a drift control sample? A: A CRM has a certified composition traceable to a national standard and is used for the initial instrument calibration. A drift control sample (or control sample) is a stable, homogeneous sample whose composition has been linked to your calibration curve. It is more affordable and robust for daily checks of instrument stability and to determine when a full recalibration is necessary [72].
Q: Can I just use a piece of bar stock as a permanent control sample? A: While possible, it is not recommended for quality-assured work. Official guidelines (e.g., DIN 51008-2) state that control samples must be comparable in precision to recalibration samples. A proper control sample should be compositionally stable, homogeneous, and its values statistically linked to your original calibration with CRMs [72].
Q: How often should I check for drift using a control sample? A: The frequency depends on your instrument stability and measurement requirements. Standard practice includes checking [72]:
- At regular intervals (e.g., after every 100 analytical samples).
- Whenever you have reason to doubt a result.
- As part of statistical process control for quality assurance.
Q: Does a dual-beam spectrophotometer design help with drift? A: Yes. A dual-beam instrument splits the light, measuring the sample and a reference simultaneously. This design reduces signal drift and improves stability for longer measurements compared to a single-beam instrument [75].
Quantitative Data from Field and Laboratory Studies
The following tables summarize key quantitative findings on drift behavior and correction from recent research.
Table 1: Performance of Drift Correction Method for a NaI(Tl) Radioactivity Sensor in Different Environments. This study demonstrates the effectiveness of a combined gain adjustment and spectrum processing method for correcting temperature-induced drift [74].
| Experiment Environment | Temperature Range | Peak Position Channel Drift (Before Correction) | Peak Position Channel Drift (After Correction) |
|---|---|---|---|
| Laboratory Air | -5°C to 50°C | Not Specified | Within ±2 channels |
| Laboratory Water | -5°C to 50°C | Not Specified | Within ±1 channel |
| Offshore Seawater Site | Field Conditions | Significant drift observed | Effectively corrected; met long-term operation requirements |
Table 2: Common Symptoms and Affected Elements from Spectrometer Subsystem Failures. Data derived from troubleshooting common Optical Emission Spectrometer (OES) issues [3].
| Problem Area | Key Symptom | Elements Typically Affected |
|---|---|---|
| Vacuum Pump | Constant low readings; pump is hot, loud, or leaking oil. | Carbon (C), Phosphorus (P), Sulfur (S), Nitrogen (N) – all low-wavelength elements [3]. |
| Contaminated Argon | A burn that appears white or milky; inconsistent or unstable results. | All elements, as the machine analyzes both the material and the contamination [3]. |
| Dirty Windows | Analysis drift requiring frequent recalibration; poor analysis reading. | All elements, as signal intensity is reduced [3]. |
Experimental Protocol: Vicarious Field Calibration
This protocol is essential for validating the radiometric accuracy of imaging spectrometers in the field, as practiced by the NEON program [73].
Objective: To verify lab-determined calibration parameters and radiometric accuracy using a known, ground-truthed target. Principle: The method involves flying the airborne spectrometer over a large, uniform calibration target with a pre-measured reflectance. Simultaneous ground-truth measurements are taken with a field spectrometer to account for atmospheric effects.
Materials:
- Airborne imaging spectrometer (e.g., NEON NIS)
- Large, certified reflectance tarps (e.g., 48% and 3% reflectance)
- Field spectrometer (e.g., ASD field spectrometer)
- GPS for precise location logging
- Radiometrically and spectrally calibrated light source (for pre-/post-flight lab checks)
Procedure:
- Pre-flight Lab Calibration: The imaging spectrometer is spectrally and radiometrically calibrated in a lab using a NIST-traceable light source before the flight season [73].
- Site Setup: Lay out the large calibration tarps on a flat, open area free of obstructions.
- Ground-Truthing: Simultaneously with the aircraft overpass, use the field spectrometer to measure the reflectance of the tarp at multiple points. This provides the "true" reflectance value [73].
- Aerial Data Collection: Fly the imaging spectrometer over the tarp site, ensuring the tarp fills a sufficient number of pixels.
- Data Processing:
- Convert the raw sensor data (Digital Numbers) to at-sensor radiance using the lab calibration parameters.
- Apply an atmospheric correction algorithm to the at-sensor radiance data, using the ground-truthed tarp reflectance as a reference point to derive accurate surface reflectance [73].
- Validation: Compare the reflectance derived from the airborne sensor with the ground-truthed measurements. The difference indicates the instrument's performance and any potential drift.
This workflow is depicted in the following diagram:
The Scientist's Toolkit: Essential Research Reagents & Materials
This table lists key materials required for conducting drift monitoring and calibration in spectrometer research.
Table 3: Essential Materials for Drift Monitoring and Calibration
| Item Name | Function & Explanation |
|---|---|
| Drift Monitors / Control Samples | Stable, homogeneous samples with known composition used for routine checks of instrument stability. They are more affordable than CRMs and are essential for detecting when an instrument begins to drift [20] [72]. |
| Certified Reference Materials (CRMs) | Materials with a certified composition, traceable to national standards. CRMs are indispensable for the initial calibration of the spectrometer, establishing the accurate baseline against which all samples are measured [72]. |
| NIST-Traceable Calibration Light Sources | Light sources (e.g., irradiance lamps, wavelength calibration lamps like Hg-Ar) with an output calibrated traceable to NIST. They are used for absolute radiometric and wavelength calibration of the spectrometer system [4] [73]. |
| Calibration Tarps | Large panels with a known and stable reflectance factor. Used in vicarious calibration of airborne imaging spectrometers to validate radiometric accuracy under real-world conditions [73]. |
| On-Board Calibration (OBC) System | An integrated system within the spectrometer that may include a dark shutter, broadband light source, and laser. Used for automatic pre- and post-flight checks to monitor the instrument's radiometric stability over time [73]. |
Assessing Model Transferability and Long-term Stability Across Instrument Platforms
Frequently Asked Questions (FAQs)
Q1: What is model transferability in spectroscopy, and why is it important? Model transferability refers to the ability to successfully use a calibration model developed on one spectrometer (the "master" instrument) on other instruments of the same type without significant loss of predictive performance [76]. This is crucial for deploying multiple spectrometers in the field, as it eliminates the need to develop individual calibration models for each instrument, saving significant time and resources [76].
Q2: What are the common causes of long-term instrumental drift? Long-term instrumental drift in techniques like Gas Chromatography-Mass Spectrometry (GC-MS) can be caused by several factors, including instrument power cycling, column replacement, mass spectrometer tuning, ion source cleaning, filament replacement, and quadrupole cleaning [7]. These factors can alter or attenuate chromatographic and mass spectrometric signals over time, affecting data reliability.
Q3: Can calibration models be directly transferred between portable spectrometers? Yes, direct model transferability has been demonstrated as feasible with modern, robust spectrometer designs. Studies using MicroNIR spectrometers have shown high cross-unit prediction success rates for both classification (e.g., polymer identification) and quantification (e.g., active pharmaceutical ingredients) tasks without applying additional calibration transfer techniques [76].
Q4: How does environmental humidity affect the stability of spectroscopic measurements? Environmental factors like water vapor can introduce substantial biases in measurements. For instance, in methane isotopic composition analysis, spectral interference from water vapor is a significant challenge. This requires the application of empirical correction functions, which can be linear or quadratic, to remove humidity-induced biases and obtain accurate data [15].
Q5: What is the role of Quality Control (QC) samples in managing long-term drift? QC samples, measured periodically over time, are essential for detecting and correcting long-term instrumental drift. By establishing a correction algorithm based on the QC data, the drift observed in actual samples can be mathematically normalized, enabling reliable quantitative comparisons over extended periods [7].
Troubleshooting Guides
Issue 1: Poor Model Performance on a Secondary Instrument
Problem: A calibration model developed on a master instrument performs poorly when used on a secondary instrument, leading to inaccurate predictions.
Solution:
- Step 1: Verify Instrument Performance and Calibration. Ensure the secondary instrument is properly calibrated and maintained according to the manufacturer's specifications. Check for any hardware issues.
- Step 2: Preprocess Spectra. Apply spectral preprocessing techniques, such as scatter correction or derivatives, to minimize baseline shifts and other non-chemical variations between instruments. Research has shown that preprocessed spectra from the same sample collected by different robust spectrometers can be very similar, enabling direct transferability [76].
- Step 3: Consider a Global Model. If multiple instruments are available, pool calibration data from at least two or three instruments to develop a more robust global model that accounts for inter-instrument variations from the outset [76].
- Step 4: Apply Calibration Transfer. If direct transfer fails, use calibration transfer techniques like Piecewise Direct Standardization (PDS). This requires a small set of transfer samples measured on both the master and secondary instruments to mathematically transform the spectra from the secondary instrument to match the master's characteristics [76].
Issue 2: Significant Data Drift Over a Long-Term Study
Problem: Analytical results from the same sample change over the course of a long study (e.g., months or years), even when using the same instrument, due to instrumental drift.
Solution:
- Step 1: Implement a QC Schedule. Regularly analyze a consistent QC sample (e.g., a pooled sample from your study) throughout your experimental timeline. For a study over 155 days, 20 QC measurements were used to establish a reliable correction model [7].
- Step 2: Choose a Correction Algorithm. Apply a mathematical algorithm to model the drift observed in the QC samples. The following table compares three algorithms evaluated for correcting GC-MS data over 155 days [7]:
| Algorithm | Description | Best For | Performance Notes |
|---|---|---|---|
| Random Forest (RF) | An ensemble learning method that uses multiple decision trees. | Long-term, highly variable data. | Most stable and reliable correction model [7]. |
| Support Vector Regression (SVR) | A variant of Support Vector Machines for predicting continuous values. | Various drift patterns. | Can over-fit and over-correct data with large variations [7]. |
| Spline Interpolation (SC) | Uses segmented polynomials (e.g., Gaussian functions) to interpolate between data points. | Simpler drift patterns. | Exhibited the lowest stability in long-term correction [7]. |
- Step 3: Classify and Correct Sample Components. Categorize components in your samples for targeted correction [7]:
- Category 1 (in QC and sample): Correct using the specific model,
f<sub>k</sub>(p, t), derived for that component from QC data. - Category 2 (in sample, not in QC, but RT matches a QC peak): Correct using the model for the QC component with the adjacent retention time.
- Category 3 (in sample, not in QC, no RT match): Apply an average correction factor derived from all QC components.
- Category 1 (in QC and sample): Correct using the specific model,
- Step 4: Apply the Correction. Use the derived correction function,
f<sub>k</sub>(p, t), and the sample's batch numberpand injection ordertto calculate a correction factor,y. The corrected peak area,x'<sub>S,k</sub>, is then calculated as:x'<sub>S,k</sub> = x<sub>S,k</sub> / y[7].
Issue 3: Inaccurate Results with Portable Spectrometers on Complex Mixtures
Problem: A portable spectrometer fails to accurately identify or quantify a target analyte in a complex mixture, such as detecting a low-concentration drug in a pill.
Solution:
- Step 1: Confirm Instrument Limitations. Be aware that portable instruments often have lower resolution and sensitivity than benchtop systems. For example, Raman and IR spectroscopy may struggle to detect components present at very low concentrations (e.g., 1% fentanyl in acetaminophen) [77].
- Step 2: Use a Multi-Technique Toolkit. Combine data from multiple portable techniques to improve reliability. A study found that using at least two portable techniques (e.g., Raman, FT-IR, and mass spectrometry) allowed for the detection of all target pharmaceutical ingredients [77].
- Step 3: Perform Sample Preparation. For trace analysis, employ simple field extraction methods to concentrate the target analyte and remove interferents. For example, a portable solvent extraction tool can concentrate fentanyl from a pill matrix before IR analysis, significantly improving detection [77].
- Step 4: Leverage Advanced Data Processing. Use digital signal processing and machine learning techniques to unmask the spectral signatures of components hidden by the dominant signals in a mixture [77].
Experimental Protocols
Protocol 1: Evaluating Direct Model Transferability for Classification
This protocol is adapted from a study on polymer classification using MicroNIR spectrometers [76].
- 1. Objective: To assess whether a classification model built on one spectrometer can directly classify samples measured on other spectrometers without modification.
- 2. Materials:
- At least three spectrometers of the same model.
- Multiple physical samples (kits) representing all classes of interest (e.g., 46 polymer types).
- 3. Procedure:
- Data Collection: For each spectrometer, measure each sample from multiple kits at several different locations and orientations to account for physical variations. Collect numerous replicate scans.
- Model Building: Use data from one resin kit measured on one spectrometer (Unit 1) to build a classification model. Test several algorithms (e.g., PLS-DA, SIMCA, SVM).
- Performance Evaluation: Test the model under different conditions:
- Same-Unit-Same-Kit: Predict different spots on the same kit measured with the same unit (baseline performance).
- Cross-Unit-Same-Kit: Predict the same kit measured on a different unit (Unit 2 or 3).
- Cross-Unit-Cross-Kit: Predict a different physical kit measured on a different unit.
- 4. Analysis: Calculate prediction success rates for each scenario. High success rates in cross-unit and cross-kit tests indicate strong direct model transferability [76].
Protocol 2: Correcting Long-Term Drift in GC-MS Using QC Samples and Machine Learning
This protocol is based on a 155-day GC-MS stability study [7].
- 1. Objective: To establish a reliable procedure for correcting long-term instrumental drift in quantitative data.
- 2. Materials:
- GC-MS instrument.
- Pooled Quality Control (QC) sample that contains as many of the target analytes as possible.
- Test samples.
- 3. Procedure:
- Experimental Design: Conduct repeated measurements of test samples and QC samples over an extended period (e.g., 155 days). Measure the QC sample at regular intervals (e.g., 20 times total).
- Data Parameterization: For every measurement (QC and samples), assign two indices:
- Batch Number (
p): An integer incremented each time the instrument is turned off and on again. - Injection Order Number (
t): An integer representing the sequence of injection within a batch.
- Batch Number (
- Virtual QC Creation: Create a "virtual QC" by taking the median peak area for each component across all 20 QC measurements. This median value,
X<sub>T,k</sub>, is the reference "true" value. - Correction Factor Calculation: For each component
kin each of theiQC measurements, calculate a correction factor:y<sub>i,k</sub> = X<sub>i,k</sub> / X<sub>T,k</sub>. - Model Building: Use machine learning (Random Forest is recommended) to find the function
y<sub>k</sub> = f<sub>k</sub>(p, t)that predicts the correction factor based on the batch and injection order.
- 4. Analysis: Apply the correction model to the actual sample data. Validate the correction by checking the reduction in variability of QC measurements over time using principal component analysis (PCA) and standard deviation analysis [7].
Workflow Diagrams
Long-Term Drift Correction Workflow
Model Transferability Assessment
The Scientist's Toolkit: Essential Research Reagents and Materials
| Item | Function | Application Context |
|---|---|---|
| Pooled Quality Control (QC) Sample | A composite sample containing target analytes, used to monitor and correct for instrumental drift over time [7]. | Long-term stability studies. |
| Nafion Dryer | Removes water vapor from air samples to prevent spectral interference and humidity-induced biases in gas-phase measurements [15]. | Spectroscopic analysis of atmospheric gases (e.g., δ¹³CH₄). |
| MicroNIR Spectrometer | A miniature, robust near-infrared spectrometer noted in research for its good direct model transferability between units [76]. | Deploying multiple spectrometers for field applications. |
| SEC Column (e.g., UHPLC protein BEH SEC) | Separates protein monomers from aggregates (high-molecular-weight species) by size exclusion chromatography (SEC) [78]. | Stability studies of biotherapeutics. |
| Portable FT-IR Spectrometer | Provides molecular fingerprinting for chemical identification in the field. Often used in a toolkit with other portable techniques [77]. | On-site drug testing and material identification. |
| Support Vector Regression (SVR) | A machine learning algorithm used to model and correct for non-linear instrumental drift using data from QC samples [7]. | Algorithmic drift correction. |
Conclusion
Effective calibration drift correction is not merely a technical necessity but a fundamental requirement for reliable spectroscopic data in long-term biomedical research. By integrating robust methodological approaches—from traditional standardization to advanced machine learning—with proactive maintenance schedules and rigorous validation protocols, researchers can significantly enhance data quality and instrument longevity. Future advancements will likely focus on physics-informed neural networks, enhanced domain adaptation techniques, and automated real-time correction systems that further reduce the need for manual intervention. Embracing these strategies ensures that field spectrometers remain precise tools for critical applications in drug development and clinical research, ultimately supporting more accurate scientific conclusions and regulatory decisions.
References
- 1. How Calibration Drift Causes Errors [https://www.srpcontrol.com/how-calibration-drift-causes-errors/]
- 2. In-situ drift correction method for deep-sea optical π-FBG ... [https://www.sciencedirect.com/science/article/abs/pii/S0263224125016513]
- 3. Troubleshooting Common Spectrometer Issues [https://verichek.net/troubleshooting-common-spectrometer-issues.html]
- 4. Calibration FAQs [https://www.oceanoptics.com/faq/calibration-faqs/]
- 5. Temporal drift in calibration of Ir-192 brachytherapy sources [https://www.sciencedirect.com/science/article/pii/S240563162500106X]
- 6. Dealing with Color Drift? It Could Be Your Spectrophotometer [https://www.xrite.com/blog/spectrophotometer-color-drift]
- 7. Correction of gas chromatography–mass spectrometry long ... [https://www.nature.com/articles/s41598-025-24794-y]
- 8. Best practice methods for reliable results from your DRIFTS ... [https://specac.com/ftir-applications/best-practice-methods-for-reliable-results-from-your-drifts-setup/]
- 9. Environmental Variation or Instrumental Drift? A Probabilistic Approach to Gas Sensor Drift Modeling and Evaluation This research project has received funding from Eurostars project E115482 (CitySense). We would also like to acknowledge Senseair for the helpful technical discussions and Empa and Decentlab for providing the dataset. [https://arxiv.org/html/2406.17488v1]
- 10. Calibration Update and Drift Correction for Electronic ... [https://www.frontiersin.org/journals/chemistry/articles/10.3389/fchem.2018.00433/full]
- 11. A drift correction optimization technique for the reduction of ... [https://www.sciencedirect.com/science/article/abs/pii/S0584854714000858]
- 12. The Importance of Drift Monitors and XRF Spectrometer ... [https://www.azom.com/article.aspx?ArticleID=17600]
- 13. Calibration Transfer, Part II: The Instrumentation Aspects [https://www.spectroscopyonline.com/view/calibration-transfer-part-ii-instrumentation-aspects]
- 14. Universal Calibration: Can Models Travel Successfully ... [https://www.spectroscopyonline.com/view/universal-calibration-can-models-travel-successfully-across-instruments-]
- 15. Evaluation of Calibration Strategies for Accurate δ 13 CH 4 ... [https://egusphere.copernicus.org/preprints/2025/egusphere-2025-5569/]
- 16. A 30-month field evaluation of low-cost CO2 sensors using a ... [https://amt.copernicus.org/articles/18/4871/2025/amt-18-4871-2025.html]
- 17. Spectrophotometer Troubleshooting [https://www.hinotek.com/spectrophotometer-troubleshooting/]
- 18. Spectrophotometer Selection and Troubleshooting [https://sperdirect.com/blogs/news/spectrophotometer-selection-and-troubleshooting-a-practical-guide?srsltid=AfmBOorHgHBAxtHS5-TXKP6mEcn_unEpXPeLM2ClHK_8LgDTUCO3C-aL]
- 19. Noise and performance study of sealed MRPC detectors ... [https://www.sciencedirect.com/science/article/abs/pii/S0168900225007041]
- 20. What is the Importance of Drift Monitors in Spectrometer ... [https://www.azom.com/article.aspx?ArticleID=23103]
- 21. Electronic Nose Sensor Drift Affects Diagnostic Reliability ... [https://www.mdpi.com/1424-8220/22/23/9246]
- 22. On the Validity and Benefit of Manual and Automated Drift ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12101172/]
- 23. Correction of field instabilities in biomolecular solid-state ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10539777/]
- 24. Evaluation and correction for long-term drift of hydraulic ... [https://www.sciencedirect.com/science/article/abs/pii/S0263224118309886]
- 25. Evaluation of two methods to eliminate the effect of water ... [https://www.sciencedirect.com/science/article/abs/pii/S0016706117302628]
- 26. Initial Drift Correction and Spectral Calibration of ... [https://www.mdpi.com/2072-4292/14/23/5964]
- 27. Radiometric Calibration with a Field Spectroradiometer [https://spectralevolution.com/application-notes-miscellaneous/radiometric-calibration-with-a-field-spectroradiometer/]
- 28. Correction of gas chromatography–mass spectrometry long ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12630760/]
- 29. Correction of gas chromatography-mass spectrometry long ... [https://pubmed.ncbi.nlm.nih.gov/41257993/]
- 30. The influence of calibration on bias in quality control and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11334199/]
- 31. Measurement Traceability: Complying with ISO 17025 ... [https://www.isobudgets.com/measurement-traceability-complying-iso-17025-requirements/]
- 32. Particle Size Standards and Count Controls [https://www.applied-microspheres.com/products/particle-size-and-count-controls]
- 33. Spectrophotometer Best Practices [https://www.hunterlab.com/blog/spectrophotometer-best-practices/]
- 34. Support Vector Regression Tutorial for Machine Learning [https://www.analyticsvidhya.com/blog/2020/03/support-vector-regression-tutorial-for-machine-learning/]
- 35. Support Vector Regression (SVR) Simplified & How To ... [https://spotintelligence.com/2024/05/08/support-vector-regression-svr/]
- 36. Calibration drift in regression and machine learning ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6080675/]
- 37. Support Vector Regression (SVR) using Linear and Non- ... [https://www.geeksforgeeks.org/machine-learning/support-vector-regression-svr-using-linear-and-non-linear-kernels-in-scikit-learn/]
- 38. Visualization, Quantification, and Alignment of Spectral Drift in ... [https://pubmed.ncbi.nlm.nih.gov/28208263/]
- 39. Support Vector Regression (SVR) using linear and non- ... [https://scikit-learn.org/stable/auto_examples/svm/plot_svm_regression.html]
- 40. Fault Diagnostics Based on the Analysis of Probability ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10857158/]
- 41. Calibration Update and Drift Correction for Electronic Noses ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6167416/]
- 42. On the use of drift correction for electrochemical ... [https://www.sciencedirect.com/science/article/abs/pii/S0013468623001469]
- 43. Drift correction in multivariate calibration models using on- ... [https://www.sciencedirect.com/science/article/abs/pii/S0003267008021934]
- 44. TinyML-Based Real-Time Drift Compensation for Gas ... [https://www.mdpi.com/2227-9040/13/7/223]
- 45. Troubleshooting Common CEM Calibration Issues - Environics [https://www.environics.com/2025/05/21/troubleshooting-common-cem-calibration-issues-tips-for-field-techs/]
- 46. The Importance of Spectrometer Calibration [https://www.xrfscientific.com/importance-spectrometer-calibration/]
- 47. Strategic Use of Calibration Transfer to Minimize ... [https://www.sciencedirect.com/science/article/abs/pii/S0731708525005965]
- 48. Troubleshooting Common Spectrometer Issues [https://nelabsystems.com/troubleshooting-common-spectrometer-issues/]
- 49. Spectrophotometer Selection and Troubleshooting [https://sperdirect.com/blogs/news/spectrophotometer-selection-and-troubleshooting-a-practical-guide?srsltid=AfmBOooOAhryOZUCGIsKxOkreOTDti0gtFbn4L6w2vdEpfIRrzVtSU5e]
- 50. Automated Calibration for Rapid Optical Spectroscopy ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11590107/]
- 51. Enhancing Spectroscopic Experiment Calibration through ... [https://www.mdpi.com/2410-3896/9/2/26]
- 52. Reduce SEM Drift: Effective Strategies [https://elementpi.com/sem-drift-reduce/]
- 53. Spectrophotometer Calibration and Validation Guide [https://aelabgroup.com/spectrophotometer-calibration-and-validation-guide/]
- 54. Why Your HPLC Baseline Drifts—And How to Stop It [https://www.sepscience.com/why-your-hplc-baseline-drifts-and-how-to-stop-it-11018]
- 55. Essentials of LC Troubleshooting, Part IV: What Is Going ... [https://www.chromatographyonline.com/view/essentials-of-lc-troubleshooting-part-iv-what-is-going-on-with-the-baseline-]
- 56. Field Spectroscopy - Part One: Mitigating the impact of sub ... [https://www.malvernpanalytical.com/en/learn/knowledge-center/insights/field-spectroscopy-part-one-mitigating-the-impact-of-sub-optimal-and-changing-weather-conditions]
- 57. Flat Field Correction of a Spatial Heterodyne Spectrometer [https://www.azom.com/article.aspx?ArticleID=14055]
- 58. What Is Proactive Maintenance? | Learning Center [https://www.getmaintainx.com/learning-center/what-is-proactive-maintenance]
- 59. Proactive Maintenance [https://www.fieldex.com/en/glossary/proactive-maintenance]
- 60. Proactive Maintenance Examples, Types and Strategies ... [https://www.xenia.team/articles/proactive-maintenance-examples]
- 61. Spectrophotometer Selection and Troubleshooting [https://sperdirect.com/blogs/news/spectrophotometer-selection-and-troubleshooting-a-practical-guide?srsltid=AfmBOopWo9UcC6yJYAn-B2eaUWQq9uxikxSmnrRVaAIbyoKcbJ0JkCn3]
- 62. Calibration Science, Part VI: Validation [https://www.cannabissciencetech.com/view/calibration-science-part-vi-validation]
- 63. Calibration Transfer, Part III: The Mathematical Aspects [https://www.spectroscopyonline.com/view/calibration-transfer-part-iii-mathematical-aspects]
- 64. Root Mean Square Error of Prediction - an overview [https://www.sciencedirect.com/topics/engineering/root-mean-square-error-of-prediction]
- 65. MSE und RMSE - Error estimators in NIR spectroscopy [https://www.en.silicann.com/blog/post/mse-und-rmse-error-estimators-in-nir-spectroscopy/]
- 66. Effects of Environmental Factors on the Performance of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12526864/]
- 67. Calibration Practices in Clinical Mass Spectrometry [https://pmc.ncbi.nlm.nih.gov/articles/PMC9467832/]
- 68. Calibration Transfer, Part VI: The Mathematics of ... [https://www.spectroscopyonline.com/view/calibration-transfer-part-vi-mathematics-photometric-standards-used-spectroscopy]
- 69. Calibration Update and Drift Correction for Electronic ... [https://pubmed.ncbi.nlm.nih.gov/30320065/]
- 70. Real-Time Correction and Long-Term Drift Compensation ... [https://www.mdpi.com/2072-666X/16/9/991]
- 71. Calibration of sea ice drift forecasts using random forest ... - TC [https://tc.copernicus.org/articles/15/3989/2021/]
- 72. Calibrating OES: Your questions answered [https://hha.hitachi-hightech.com/en/blogs-events/blogs/2020/08/24/cast-iron-webinar-q-and-a/]
- 73. Imaging Spectrometer [https://www.neonscience.org/data-collection/imaging-spectrometer]
- 74. A New Method for the Correction of Spectrum Drift Caused ... [https://www.mdpi.com/2077-1312/12/4/546]
- 75. Spectrophotometer Selection and Troubleshooting [https://sperdirect.com/blogs/news/spectrophotometer-selection-and-troubleshooting-a-practical-guide?srsltid=AfmBOoo2FoE4QIdvZr8YS0Ou-O_mwFg7N-TzhQyMWYazK-7hjYxrtD2R]
- 76. Investigation of Direct Model Transferability Using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6571657/]
- 77. Portable Spectrometers Give On-Site Drug Testing a Boost [https://pmc.ncbi.nlm.nih.gov/articles/PMC9951272/]
- 78. Simplified kinetic modeling for predicting the stability of ... [https://www.nature.com/articles/s41598-025-07037-y]