Electron Energy Level Transitions in Spectroscopy: From Fundamental Principles to Advanced Biomedical Applications
This article provides a comprehensive exploration of electron energy level transitions, the fundamental process underlying modern spectroscopic analysis.
Electron Energy Level Transitions in Spectroscopy: From Fundamental Principles to Advanced Biomedical Applications
Abstract
This article provides a comprehensive exploration of electron energy level transitions, the fundamental process underlying modern spectroscopic analysis. Tailored for researchers, scientists, and drug development professionals, it details the core quantum mechanical principles governing how electrons absorb and emit energy. The scope extends from foundational concepts and selection rules to advanced methodological applications in pharmaceutical analysis and medical imaging, including radiotheranostics and photodetector design. It further addresses critical challenges in detecting weak transitions and optimizing signals, while covering rigorous protocols for data validation and comparative analysis of spectroscopic techniques. This guide serves as an essential resource for leveraging spectroscopic data to drive innovation in biomedical research and clinical diagnostics.
Quantum Foundations: Understanding How Electrons Interact with Light
The principle of energy quantization represents a foundational pillar of modern physics and chemistry, marking a radical departure from classical theories that described energy as a continuous variable. This concept asserts that energy, particularly within atomic and molecular systems, can only exist in discrete, specific amounts or "quanta." The transition from this fundamental understanding to its practical application in spectroscopic techniques forms the bedrock of modern analytical science, with profound implications across chemical analysis, pharmaceutical development, and materials science.
The quantized nature of energy manifests most directly in the behavior of electrons within atoms and molecules. These electrons are restricted to specific energy levels, often visualized as orbits or states. The lowest possible energy state for an electron is termed the ground state, representing its most stable configuration. When an electron absorbs a precise quantum of energy, it can transition to a higher-energy, less stable configuration known as an excited state. The existence of these discrete states and the specific energies required for transitions between them provide the theoretical framework for interpreting atomic and molecular spectra, which serve as unique fingerprints for chemical identification [1] [2].
Fundamental Principles of Quantum States
Defining Ground and Excited States
In an atom, electrons tend to adopt an arrangement that minimizes the atom's total energy. This lowest energy condition is known as the ground state [1]. It is the default, stable state of an atom under normal conditions. However, when atoms absorb energy from an external source—such as heat or electrical discharge—their electrons can gain energy and move to higher energy levels. These higher-energy configurations are called excited states [1]. An atom in an excited state is inherently unstable; its electrons will eventually release their excess energy and return to lower energy states or the ground state [1] [2].
The Quantum Mechanical Framework
The quantum revolution, pioneered by Max Planck and Albert Einstein, introduced the concept that energy transfer at the atomic scale occurs in discrete packets, or quanta. Planck's work on blackbody radiation revealed that the energy (E) of an oscillator is quantized and proportional to its frequency (f), as described by the equation (E = hf), where (h) is Planck's constant ((6.626 \times 10^{-34} \text{J·s})) [3] [4]. Einstein extended this idea to light itself, proposing that electromagnetic radiation consists of quantized particles called photons, each carrying energy (E = hf) [4]. This particle-like nature of light was confirmed through explanations of the photoelectric effect, where light below a certain frequency cannot eject electrons from a metal surface, regardless of intensity [4].
For electrons confined within atoms, this quantization means they can only occupy specific energy levels, much like a person can only stand on specific rungs of a ladder and not between them [3] [5]. The energy levels are not equally spaced; they become closer together as the principal quantum number (n) increases [5]. An electron's transition between these allowed levels involves the absorption or emission of a photon with an energy exactly equal to the energy difference between the two states: (\Delta E = E{\text{final}} - E{\text{initial}} = hf) [1] [2] [5].
Electronic Transitions and Spectroscopy
Molecular Orbitals and Chromophores
In molecular systems, the concept of atomic energy levels expands into molecular orbitals. Electrons reside in bonding orbitals (lower energy) or anti-bonding orbitals (higher energy) in the ground state. The Highest Occupied Molecular Orbital (HOMO) is the highest-energy orbital containing electrons, while the Lowest Unoccupied Molecular Orbital (LUMO) is the lowest-energy vacant orbital [6]. Electronic transitions often involve the promotion of an electron from the HOMO to the LUMO, and the energy difference between these orbitals determines the wavelength of light absorbed [6].
Chromophores are the light-absorbing molecular components responsible for color, typically featuring groups like C=C, C=O, or aromatic rings [6]. Conjugation—the alternation of single and multiple bonds—delocalizes electrons across a larger part of the molecule, which reduces the HOMO-LUMO energy gap. This results in absorption at longer wavelengths, a phenomenon known as a bathochromic shift or red shift [6]. For example, beta-carotene, a highly conjugated polyene, absorbs visible light and appears orange [6].
Table 1: Common Chromophores and Their Transitions
| Chromophore | Example Compound | Transition Type | Typical Absorption Range (nm) |
|---|---|---|---|
| C=C (Isolated) | Ethene | π → π* | ~170 |
| C=C (Conjugated) | 1,3-Butadiene | π → π* | ~220 |
| C=O | Acetone | n → π* | ~280 |
| Aromatic Ring | Benzene | π → π* | ~260 |
| Extended Conjugation | Beta-Carotene | π → π* | ~450 (Visible) |
Auxochromes are functional groups (e.g., -OH, -NH₂) that lack inherent color but can modify the absorption of a chromophore when attached. Typically containing lone pairs of electrons, auxochromes can donate electrons to the chromophore, often leading to a bathochromic shift and increased absorption intensity (a hyperchromic effect) [6]. Solvent polarity and pH can also induce spectral shifts by stabilizing or destabilizing the ground or excited states [6].
Absorption Spectroscopy and the Beer-Lambert Law
Absorption spectroscopy measures the extent to which a sample absorbs light at different wavelengths. The Beer-Lambert Law quantifies this relationship, providing a foundation for quantitative analysis [6]. The law is expressed as: [ A = \epsilon b c ] where:
- (A) is the measured absorbance (unitless),
- (\epsilon) is the molar absorptivity or extinction coefficient (L·mol⁻¹·cm⁻¹),
- (b) is the path length of the light through the sample (cm),
- (c) is the concentration of the absorbing species (mol·L⁻¹) [6].
Absorbance is directly proportional to concentration for dilute solutions, enabling the determination of unknown concentrations via calibration curves. Deviations from linearity can occur at high concentrations due to molecular interactions [6]. The molar absorptivity ((\epsilon)) reflects the probability of an electronic transition. "Allowed" transitions, which obey selection rules, have high (\epsilon) values (typically >10,000 L·mol⁻¹·cm⁻¹), whereas "forbidden" transitions have low (\epsilon) values (<100 L·mol⁻¹·cm⁻¹) [6].
Table 2: Characteristics of Electronic Transitions
| Transition Type | Orbitals Involved | Typical Molar Absorptivity (ε, L·mol⁻¹·cm⁻¹) | Example Chromophore |
|---|---|---|---|
| π → π* | Bonding π to Anti-bonding π* | High (5,000 - 25,000) | Alkenes, Conjugated systems |
| n → π* | Non-bonding to Anti-bonding π* | Low (10 - 100) | Carbonyl (C=O) |
| σ → σ* | Bonding σ to Anti-bonding σ* | High | Alkanes |
| n → σ* | Non-bonding to Anti-bonding σ* | Medium | Alcohols, Amines |
| Charge Transfer | Electron Donor to Acceptor | Very High (> 20,000) | Metal-Ligand Complexes |
Experimental Methodologies in Spectroscopy
Generating and Analyzing Atomic Emission Spectra
Experimental Protocol: Observation of the Hydrogen Emission Spectrum
Principle: When atoms are excited by an energy source (heat or electricity), their electrons jump to higher energy levels. Upon returning to lower levels, they emit photons of specific energies, producing a unique line spectrum—a series of bright lines at discrete wavelengths against a dark background [2] [7]. This contrasts with a continuous spectrum, which contains an unbroken sequence of colors over a broad range, produced by white light [2].
Materials and Equipment:
- Hydrogen Gas Discharge Tube: A sealed glass tube filled with pure hydrogen gas at low pressure.
- High-Voltage Power Supply: Provides the electrical energy (typically several thousand volts) to excite the hydrogen atoms.
- Diffraction Grating or Prism: An optical component to disperse the emitted light into its constituent wavelengths.
- Viewing Scope or Spectrometer: An instrument for observing and measuring the angles or positions of the spectral lines.
- Calibration Light Source: A source with known emission lines (e.g., helium or neon) for wavelength calibration.
Procedure:
- Setup: Place the hydrogen discharge tube in its holder and connect it to the high-voltage power supply, ensuring all safety precautions are followed. Position the diffraction grating or prism so that light from the tube passes through it. Arrange the viewing scope or spectrometer to capture the dispersed light.
- Excitation: Turn on the power supply. Electrical discharge will pass through the tube, causing the hydrogen gas to glow with a characteristic pinkish color.
- Observation: Look through the viewing scope. You will observe several discrete colored lines against a black background, rather than a continuous rainbow. These are the emission lines of hydrogen.
- Measurement and Identification: Use the calibrated scale in the spectrometer to measure the wavelength of each visible line. The four most prominent lines in the visible region are:
- Data Analysis: Compare the measured wavelengths to the known values for the Balmer series of hydrogen. The transitions can be verified using the Rydberg formula (see Section 5.1).
Recording a UV-Visible Absorption Spectrum
Experimental Protocol: UV-Vis Absorption Measurement of a Chromophore
Principle: Molecules containing chromophores absorb light in the UV-visible region, promoting electrons from the ground state to an excited state. The resulting absorption spectrum provides information about the molecular structure, concentration, and environment [6].
Materials and Equipment:
- UV-Visible Spectrophotometer: An instrument consisting of a light source (deuterium lamp for UV, tungsten for visible), a monochromator to select wavelengths, a sample compartment, and a detector.
- Matched Cuvettes: A pair of high-quality quartz (for UV) or glass (for visible only) cells to hold the solvent (blank) and the sample solution.
- Analytical Grade Solvent: A solvent that does not absorb significantly in the spectral region of interest (e.g., water, hexane, methanol).
- Pure Analyte: The compound to be studied.
Procedure:
- Sample Preparation: Accurately weigh a small amount of the pure analyte and dissolve it in the chosen solvent to prepare a solution of known concentration (typically in the micromolar to millimolar range). For quantitative work, prepare a series of standard solutions for a calibration curve.
- Instrument Preparation: Turn on the spectrophotometer and allow it to warm up. Set the desired wavelength range (e.g., 200-800 nm).
- Blank Measurement: Fill a cuvette with the pure solvent, place it in the sample compartment, and record a baseline spectrum (blank). This corrects for any absorption from the solvent or cuvette.
- Sample Measurement: Replace the blank cuvette with the cuvette containing your sample solution. Record the absorption spectrum across the set wavelength range.
- Data Analysis:
- Identify the wavelength of maximum absorption (( \lambda_{\text{max}} )) for each peak.
- Use the Beer-Lambert Law (( A = \epsilon b c )) to calculate the concentration of an unknown or the molar absorptivity (( \epsilon )) of the compound.
- Note the effects of conjugation, auxochromes, or solvent on the position and intensity of absorption bands.
The Scientist's Toolkit: Key Research Reagents and Materials
Table 3: Essential Materials for Spectroscopic Analysis
| Item | Function & Application |
|---|---|
| Quartz Cuvettes | Contain liquid samples for UV-Vis spectroscopy; quartz is transparent down to ~200 nm, unlike glass. |
| Hydrogen/Deuterium Discharge Lamps | Provide characteristic line spectra for element identification and instrument calibration in emission spectroscopy. |
| Deuterium and Tungsten Lamps | Broad-spectrum light sources for UV and visible regions, respectively, in UV-Vis spectrophotometers. |
| Diffraction Grating | Disperses light into its component wavelengths within a spectrometer or monochromator. |
| Standard Reference Materials | Compounds with known, stable spectral properties (e.g., holmium oxide filter for wavelength calibration in UV-Vis). |
| High-Purity Solvents | Dissolve analytes without introducing interfering absorbances (e.g., acetonitrile, hexane, water for HPLC). |
| Deuterated Solvents | Used in NMR spectroscopy to provide a lock signal and avoid overwhelming proton signals from the solvent. |
Data Analysis and Computational Applications
The Rydberg Formula and Hydrogen Spectrum
The empirical Rydberg formula accurately predicts the wavelengths of all spectral lines in the hydrogen atom. It is given by: [ \frac{1}{\lambda} = RZ^2 \left( \frac{1}{n'^{2}} - \frac{1}{n^{2}} \right) ] where:
- ( \lambda ) is the wavelength of the emitted or absorbed light,
- ( R ) is the Rydberg constant (approximately ( 1.097 \times 10^7 \, \text{m}^{-1} )),
- ( Z ) is the atomic number (1 for hydrogen),
- ( n' ) and ( n ) are positive integers representing the lower and upper energy levels, respectively, with ( n > n' ) [7].
For the hydrogen emission spectrum, different series of lines correspond to electrons falling to different lower energy levels (( n' )):
- Lyman series: ( n' = 1 ) (Ultraviolet)
- Balmer series: ( n' = 2 ) (Visible and near-UV) [7]
- Paschen series: ( n' = 3 ) (Infrared)
The Balmer series is the most famous, as its lines are within the visible region and were the first to be studied extensively [7].
Predicting Spectral Lines
The total number of possible spectral lines emitted when an electron drops from a higher energy level ( n ) to all possible lower levels is given by: [ \text{Number of Spectral Lines} = \frac{n(n-1)}{2} ] For example, an electron excited to the ( n=4 ) level can produce ( \frac{4(4-1)}{2} = 6 ) distinct spectral lines as it returns to the ground state via various pathways [7].
Advanced Applications in Research and Industry
The principles of quantized energy and electronic transitions underpin powerful analytical techniques used in diverse scientific fields.
- Pharmaceutical Development: Atomic absorption and emission spectroscopy are used to detect trace metal impurities in active pharmaceutical ingredients (APIs) and drug products, ensuring product safety and compliance with regulatory standards [7].
- Astronomical Spectroscopy: By analyzing the emission and absorption spectra of light from stars and galaxies, astronomers can determine their chemical composition and physical properties, such as temperature and relative motion. This provides direct evidence for the existence of different elements in the universe [2] [7].
- Environmental Monitoring: Atomic spectroscopy is employed to measure concentrations of heavy metals (e.g., lead, mercury, arsenic) and other pollutants in water, soil, and air samples with high sensitivity and accuracy [7].
- Metallurgy and Materials Science: Emission spectra are used for the qualitative and quantitative elemental analysis of metals and alloys, crucial for quality control and material identification [7].
- Biochemical Research: UV-Vis spectroscopy is a standard tool for quantifying biomolecules like proteins and nucleic acids, as well as for studying enzyme kinetics and ligand-binding interactions [6].
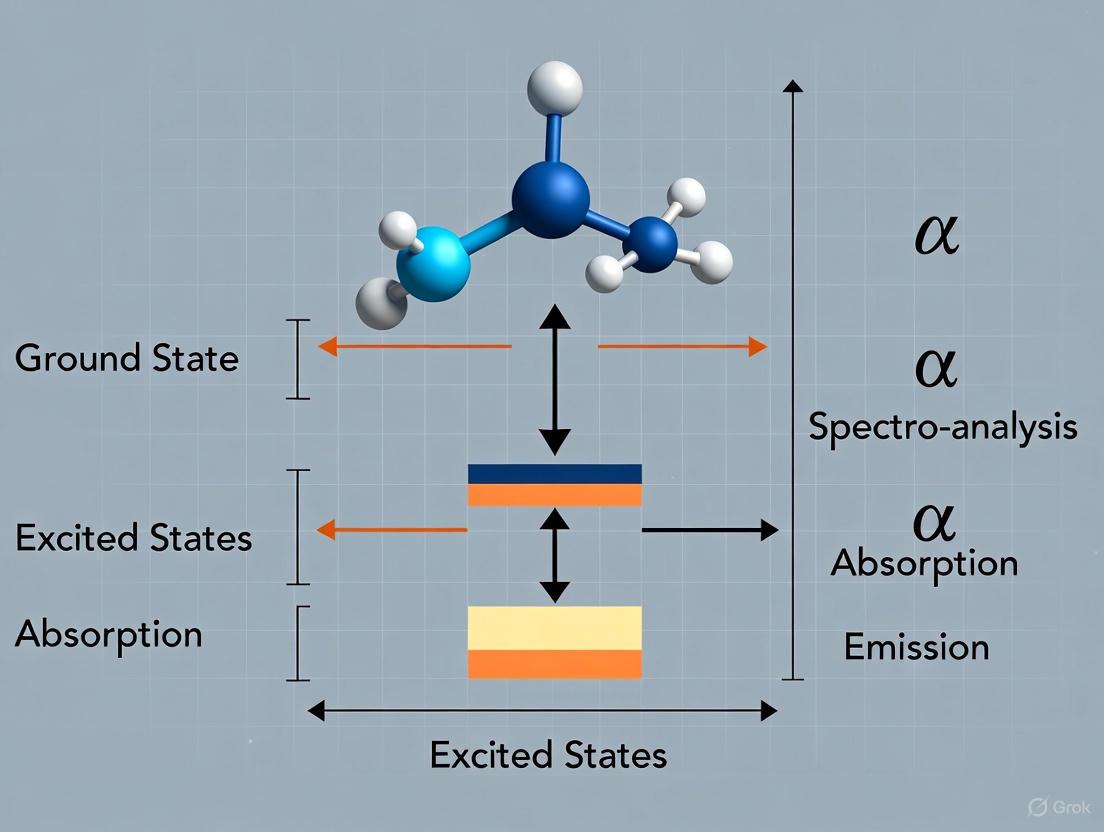
Diagram: Electronic Transitions and Spectral Formation
Diagram: Spectroscopic Experiment Workflow
Fluorescence is a form of photoluminescence that occurs when a substance absorbs electromagnetic radiation and subsequently emits light from electronically excited states. This cyclical process encompasses distinct stages: excitation of a fluorophore by photon absorption, a finite excited-state lifetime, and emission of a photon at a longer wavelength during relaxation to the ground state. The entire sequence occurs rapidly, with fluorescence lifetimes typically ranging from nanoseconds to hundreds of nanoseconds. This phenomenon provides the foundation for powerful analytical techniques utilized across chemical analysis, biological research, and drug development due to its exquisite sensitivity, specificity, and adaptability for spatial and temporal resolution.
The significance of fluorescence technology extends from fundamental research to practical applications. In drug development, fluorescence-based assays enable high-throughput screening, receptor-ligand binding studies, and cellular uptake measurements. Advanced fluorescence methodologies provide researchers with tools to investigate dynamic processes in living systems, monitor protein interactions, detect pathogens, and diagnose diseases with exceptional precision.
Fundamental Mechanisms and Jablonski Diagram
The photophysical processes underlying fluorescence are classically represented using a Jablonski energy diagram, named after Polish physicist Alexander Jablonski. This diagram illustrates the electronic states of a molecule and the transitions between them following light absorption [8].
Key Processes Visualized
The Jablonski diagram above illustrates several critical photophysical processes and energy transitions [9] [8]:
Absorption: A photon of energy hν_EX is absorbed, promoting the fluorophore from the ground state (S₀) to a higher electronic excited state (S₁, S₂) in approximately 10⁻¹⁵ seconds. This process occurs without a change in electron spin pairing.
Vibrational Relaxation: The excited fluorophore rapidly relaxes (10⁻¹² to 10⁻¹⁰ seconds) to the lowest vibrational level of the first excited singlet state (S₁), dissipating excess energy as heat through molecular collisions.
Internal Conversion: Non-radiative transition between electronic states of the same spin multiplicity (e.g., S₂ to S₁) occurs through vibrational coupling, typically within 10⁻¹² to 10⁻¹⁴ seconds.
Fluorescence Emission: The fluorophore returns to the ground state (S₀) by emitting a photon of energy hν_EM (10⁻⁹ to 10⁻⁷ seconds). The emitted photon has lower energy than the absorbed photon due to prior energy dissipation.
Intersystem Crossing: A non-radiative transition (10⁻¹⁰ to 10⁻⁸ seconds) between states of different spin multiplicity (e.g., S₁ to T₁), forming a metastable triplet state.
Phosphorescence: Emission from the triplet state (T₁) to the ground state (S₀) with much longer lifetimes (10⁻³ to 10⁰ seconds) due to the forbidden nature of this spin-flip transition.
Quantitative Fluorescence Parameters
Key Photophysical Properties
Table 1: Fundamental fluorescence parameters and their significance
| Parameter | Definition | Significance | Typical Range/Values |
|---|---|---|---|
| Extinction Coefficient (ε) | Measure of how strongly a fluorophore absorbs light at a specific wavelength | Determines brightness; higher ε means more light capture per molecule | Varies by dye; ~80,000 M⁻¹cm⁻¹ for fluorescein at 494 nm [9] |
| Quantum Yield (QY) | Ratio of photons emitted to photons absorbed | Measures fluorescence efficiency; higher QY means brighter emission | 0-1.0 (e.g., 0.93 for fluorescein, 0.79 for rhodamine B) [9] |
| Stokes Shift | Energy/wavelength difference between absorption and emission maxima | Enables separation of excitation and emission signals; reduces self-absorption | 20-100 nm for organic fluorophores [10] [11] |
| Fluorescence Lifetime (τ) | Average time a molecule spends in the excited state before emission | Sensitive to molecular environment; enables fluorescence lifetime imaging | Nanoseconds to hundreds of nanoseconds [8] |
| Dissociation Constant (K_d) | Equilibrium constant for dye-ion complex dissociation | Determines suitable ion concentration range for sensing applications | Varies by dye-ion pair; dictates measurement range [11] |
Timescales of Fluorescence Processes
Table 2: Characteristic timescales for photophysical processes in fluorescence
| Transition | Process | Timescale (Seconds) | Rate Constant |
|---|---|---|---|
| S₀ → S₁ or Sₙ | Absorption (Excitation) | 10⁻¹⁵ | Instantaneous [8] |
| Sₙ → S₁ | Internal Conversion | 10⁻¹⁴ to 10⁻¹⁰ | k_ic [8] |
| S₁ → S₁ | Vibrational Relaxation | 10⁻¹² to 10⁻¹⁰ | k_vr [8] |
| S₁ → S₀ | Fluorescence | 10⁻⁹ to 10⁻⁷ | k_f or Γ [8] |
| S₁ → T₁ | Intersystem Crossing | 10⁻¹⁰ to 10⁻⁸ | k_pT [8] |
| T₁ → S₀ | Phosphorescence | 10⁻³ to 10⁰ | k_p [8] |
Advanced Fluorescence Mechanisms and Experimental Systems
Recent research has revealed sophisticated photoluminescent behaviors that deviate from Kasha's rule, which states that emission occurs only from the lowest excited state. Excitation-wavelength-dependent (Ex-De) photoluminescence represents a significant advancement, where emission properties change according to the excitation wavelength [12]. This phenomenon enables single-molecule systems to emit different colors when excited at different wavelengths, providing opportunities for advanced applications in multiplexed sensing and anti-counterfeiting technologies.
A groundbreaking 2025 study demonstrated an organic molecule integrating both excited-state intramolecular proton transfer (ESIPT) and proton-coupled electron transfer (PCET) mechanisms, exhibiting remarkable Ex-De behavior [12]. This system achieved unprecedented absolute fluorescence quantum yields of 55.6% (λex: 396 nm) and 69.6% (λex: 363 nm) when embedded in a poly(vinyl alcohol) film, substantially higher than previously reported Ex-De organic molecules. The underlying mechanism was elucidated through transient absorption and spectroelectrochemistry spectra, revealing that varying excitation wavelengths switches the dominant process between ESIPT and PCET, creating the observed excitation-dependent emission color changes from greenish-blue to yellow-green [12].
Materials and Equipment:
- Spectrofluorometer with temperature control capability
- UV-vis spectrophotometer
- Temperature-controlled sample chamber (180-400 K range)
- Quartz cuvettes (1 cm path length)
- High-performance liquid chromatography (HPLC) system with C18 column
- Poly(vinyl alcohol) (PVA) film substrate
- Target compound in methanol solution (33.3 μM concentration)
Procedure:
- Sample Purification and Verification: First, verify compound purity using HPLC with a C18 reversed-phase column. A single peak at the retention time confirms sample purity, ensuring subsequent observations are not due to impurities [12].
Absorption Spectroscopy: Record UV-vis absorption spectra of the compound in methanol solution (50 μM concentration) across 250-500 nm range. Identify distinct absorption bands and their maxima [12].
Temperature-Dependent PL Measurements:
- Place sample in temperature-controlled chamber and set initial temperature to 300 K.
- Excite sample at 360 nm and record emission spectrum from 370-650 nm.
- Gradually decrease temperature to 180 K in 20 K increments, recording PL spectrum at each temperature.
- Generate temperature-emission mapping and calculate CIE chromaticity coordinates for each temperature [12].
Excitation-Wavelength-Dependent Measurements:
- Maintain constant temperature (e.g., 300 K).
- Record complete emission spectra while systematically varying excitation wavelength from 340-420 nm.
- Determine absolute fluorescence quantum yields at key excitation wavelengths (e.g., 363 nm and 396 nm) using integrated sphere attachment [12].
Film Preparation and Characterization:
- Embed compound in PVA film matrix.
- Repeat excitation-wavelength-dependent measurements on solid-state film.
- Evaluate room-temperature phosphorescence after cessation of excitation light [12].
Data Analysis:
- Plot normalized excitation and emission spectra for different conditions.
- Create temperature-emission intensity profile and CIE coordinate diagrams.
- Calculate quantum yields and compare emission ratios at different excitation wavelengths.
- Perform global lifetime analysis of transient absorption data to produce decay-associated difference spectra.
Research Reagent Solutions and Materials
Table 3: Essential research reagents and materials for fluorescence studies
| Reagent/Material | Function/Application | Examples/Specifications |
|---|---|---|
| ESIPT-PCET Probes | Excitation-wavelength-dependent studies | 2-(2-hydroxy-5-methylphenyl)benzothiazole derivatives with spinacine moiety [12] |
| Ion-Sensitive Dyes | Measuring intracellular ion concentrations | Fluo-3 (Ca²⁺), Fura-2 (Ca²⁺), Indo-1 (Ca²⁺); selection based on K_d matching expected concentration [11] |
| Polymer Matrix Materials | Solid-state fluorescence studies | Poly(vinyl alcohol) films for embedding fluorophores [12] |
| Reference Standards | Calibration and quantification | Fluorescent microsphere standards for microscopy/flow cytometry; fluorescent standard solutions for spectrofluorometry [9] |
| Solvent Systems | Spectroscopic studies | Anhydrous, spectroscopic-grade solvents (methanol, dichloromethane) for solution-phase studies [12] |
While light-induced fluorescence remains the most common approach, technological advancements have expanded excitation strategies to include diverse energy sources, each offering unique advantages for specialized applications [13]:
Mechanoluminescence: Utilizes mechanical energy for excitation, enabling real-time mechanical signal detection in smart devices and structural health monitoring.
X-ray-induced Luminescence: Leverages high-energy electromagnetic radiation for deep-tissue imaging, alloy element analysis, and non-destructive examination of archaeological samples.
Chemiluminescence/Bioluminescence: Employs chemical or biological energy sources, eliminating the need for external light sources and avoiding photobleaching while enabling endogenous sensing.
Electroluminescence: Applies electrical energy for excitation, forming the basis for OLED displays and energy-efficient lighting panels.
Magnetoluminescence: Uses magnetic fields for excitation, with emerging applications in tumor imaging, targeted drug delivery, and thermotherapy through magnetofluorescent nanostructures.
Experimental Workflow for Advanced Fluorescence Studies
The workflow above outlines a comprehensive approach for characterizing advanced photoluminescent systems, particularly those exhibiting excitation-wavelength-dependent behavior. This methodology integrates steady-state and time-resolved spectroscopic techniques with electrochemical analysis to elucidate complex photophysical mechanisms such as ESIPT and PCET processes [12].
The fundamental principles of absorption, excitation, and fluorescence provide a robust framework for understanding molecular photophysics and developing advanced analytical techniques. The core phenomenon of fluorescence—characterized by electronic transitions between defined energy states governed by the Jablonski diagram—remains consistent across applications, though its manifestations continue to expand through sophisticated molecular designs and excitation strategies.
Recent advances in excitation-wavelength-dependent photoluminescence represent a significant departure from traditional Kasha's rule behavior, offering new opportunities for single-molecule multiplexing and anti-counterfeiting technologies. The integration of multiple mechanisms such as ESIPT and PCET within single molecular systems, coupled with emerging excitation strategies utilizing diverse energy sources, continues to broaden the application landscape of fluorescence technology. These developments promise enhanced capabilities in drug discovery, biomedical imaging, and materials science, driven by ongoing research into the intricate relationship between molecular structure, electronic transitions, and photophysical behavior.
In the fields of electronic spectroscopy and quantum chemistry, selection rules are formal constraints that dictate the probability of a system transitioning from one quantum state to another [14]. These rules are fundamental to interpreting electronic spectra, predicting molecular behavior, and designing materials with specific optical properties. They determine whether a given transition is "allowed" (high probability and intensity) or "forbidden" (low probability and intensity) based on the symmetry properties of the initial and final states and the operator responsible for the transition [15].
The theoretical foundation for these rules lies in the transition moment integral, which quantifies the probability of an electronic transition [14]. This integral must be non-zero for a transition to be allowed. In practical terms, rather than calculating this complex integral for every possible transition, scientists determine the symmetry of the transition moment function. If this function is antisymmetric with respect to the key symmetry operations of the molecule, the transition is forbidden [14].
Selection rules originate from various symmetry considerations, including rotational invariance, inversion symmetry (parity), time-reversal invariance, and exchange symmetry [15]. The application of group theory to selection rules was pioneered by Eugene Wigner in the 1920s, starting with atomic spectroscopy and later extending to molecular and nuclear spectroscopy [15]. This guide focuses on three core concepts central to understanding electronic transitions: the Laporte selection rule, the spin selection rule, and the phenomenon of vibronic coupling that relaxes these strict rules.
The Laporte Selection Rule
Fundamental Principle and Parity
The Laporte selection rule, named after Otto Laporte who published it with William Frederick Meggers in 1925, is a powerful principle governing electronic transitions in atoms and molecules with a center of symmetry [16]. It states that electronic transitions that conserve parity are forbidden [16]. In more precise terms, for centrosymmetric systems (those with an inversion center), transitions between states of the same parity are forbidden, while transitions involving a change in parity are allowed [17] [16].
The concept of parity refers to the behavior of a quantum state under inversion through a center of symmetry. Orbitals and states are classified as:
- Gerade (g): Symmetric (even) with respect to inversion
- Ungerade (u): Antisymmetric (odd) with respect to inversion [16]
In atomic systems, s and d orbitals are gerade (g), while p and f orbitals are ungerade (u) [16]. Therefore, the Laporte rule specifically forbids s→s, p→p, d→d, and f→f transitions in centrosymmetric environments, while allowing s→p, p→d, d→f, etc. [16] [18].
Impact on Transition Metal Complexes
The Laporte rule is particularly important in the context of transition metal complexes, where it explains the weak intensity of many d-d transitions [16] [19]. In octahedral complexes, which possess a center of symmetry, all d orbitals have g symmetry, making d-d transitions Laporte-forbidden (g→g) [17] [18]. Despite this forbiddenness, such transitions are still observed because the selection rule can be relaxed through various mechanisms.
The intensity of these forbidden transitions is significantly weaker than allowed transitions. The molar absorptivity (ε) for Laporte-forbidden d-d transitions typically ranges from 1-100 L mol⁻¹ cm⁻¹, compared to 1,000-10⁶ L mol⁻¹ cm⁻¹ for fully allowed charge-transfer bands [18].
Table: Intensity of Electronic Transitions Based on Selection Rules
| Transition Type | Spin Rule | Laporte Rule | Typical εmax (L mol⁻¹ cm⁻¹) |
|---|---|---|---|
| Spin forbidden, Laporte forbidden | Violated | Violated | 10⁻³ - 1 |
| Spin allowed, Laporte forbidden | Obeyed | Violated | 1 - 100 |
| Spin allowed, Laporte allowed | Obeyed | Obeyed | 100 - 1,000 |
| Charge-transfer bands | Obeyed | Symmetry allowed | 1,000 - 10⁶ |
Tetrahedral complexes provide an interesting contrast to octahedral complexes. Because tetrahedral complexes lack a center of symmetry, the Laporte rule does not apply, and their d-d transitions are consequently more intense [16] [18]. This explains why tetrahedral complexes often exhibit stronger colors than their octahedral counterparts. For example, the octahedral complex [Co(H₂O)₆]²⁺ is pink with ε ≈ 10, while the tetrahedral complex [CoCl₄]²⁻ is deep blue with ε ≈ 600 [16].
The Spin Selection Rule
Fundamental Principle
Complementing the Laporte rule is the spin selection rule, which governs changes in spin state during electronic transitions. This rule states that the total spin quantum number (S) cannot change during an electronic transition (ΔS = 0) [17] [20]. In practical terms, this means transitions must occur between states of the same spin multiplicity [17].
The physical basis for this rule lies in the fact that electromagnetic radiation cannot directly flip electron spin [18]. The electric dipole operator, responsible for most electronic transitions, does not interact with electron spin. Therefore, the relative orientation of electron spins remains unchanged during the transition.
Implications for Electronic Spectra
The spin selection rule has significant implications for interpreting electronic spectra:
- Singlet-singlet transitions (e.g., S₀ → S₁) are spin-allowed [20]
- Singlet-triplet transitions (e.g., S₀ → T₁) are formally forbidden [20]
When both the spin and Laporte selection rules are violated, transitions become particularly weak. This combined effect explains the faint colors of certain transition metal complexes, such as octahedral Mn(II) and Fe(III) complexes, where transitions are both spin-forbidden and Laporte-forbidden [16].
Table: Examples of Spin-Forbidden Transitions in Coordination Chemistry
| Complex | Electronic Configuration | Color | Reason for Weak Color |
|---|---|---|---|
| [Mn(H₂O)₆]²⁺ | d⁵ high-spin | Pale pink | Spin-forbidden and Laporte-forbidden d-d transitions |
| [Fe(H₂O)₆]³⁺ | d⁵ high-spin | Pale violet | Spin-forbidden and Laporte-forbidden d-d transitions |
Despite being formally forbidden, spin-forbidden transitions can still be observed under certain conditions. Their intensities are typically much weaker than spin-allowed transitions, with molar absorptivities often below 1 L mol⁻¹ cm⁻¹ [18].
Vibronic Coupling: Relaxation of Selection Rules
Mechanism of Vibronic Coupling
Vibronic coupling represents a crucial mechanism through which formally forbidden transitions gain intensity. This phenomenon involves the coupling of electronic and vibrational motions within a molecule [17] [19]. Even in centrosymmetric molecules, nuclear vibrations cause temporary distortions that break the center of symmetry [16].
During these asymmetric vibrations, the molecular symmetry is temporarily lowered, allowing transitions that would be forbidden in the perfectly symmetric equilibrium geometry [17] [18]. This temporary loss of centrosymmetry enables d-d transitions to borrow intensity from allowed transitions through the mixing of electronic states of different parity [16].
Impact on Spectral Intensities
Vibronic coupling is primarily responsible for the observation of d-d transitions in octahedral complexes [16] [18]. While these transitions remain weaker than fully allowed transitions, vibronic coupling provides a pathway for them to occur with measurable intensity. The transitions occur during moments of asymmetric vibration when the center of symmetry is temporarily lost [18].
The extent of vibronic coupling depends on factors such as:
- The strength of electron-vibration interactions
- The energy separation between electronic states
- The specific vibrational modes involved
In rare-earth ions, the Laporte rule is relaxed when the ion is in a ligand field without an inversion center, causing distortion of the spherical symmetry and leading to mixing of electronic configurations of opposite parities [21]. These orbital mixtures between the rare-earth metal and the host matrix ligand make otherwise forbidden f-f transitions observable [21].
Experimental Methodologies and Technical Approaches
Spectroscopic Characterization Techniques
The investigation of selection rules and their relaxation mechanisms relies on sophisticated spectroscopic techniques:
Electronic Absorption Spectroscopy measures the attenuation of light passing through a sample, providing information about allowed and forbidden transitions through their intensities and positions [22]. Temperature-controlled studies are particularly valuable, as decreasing temperature reduces thermal broadening, allowing vibrational fine structure to emerge in electronic spectra [22].
Low-Temperature Spectroscopy is essential for resolving vibronic structure. As temperature decreases, vibrational hot bands are minimized, sharpening spectral features and revealing the vibrational progression associated with electronic transitions [22]. This enables researchers to study the Franck-Condon principle, which states that electronic transitions occur much faster than nuclear motions, resulting in vertical transitions on potential energy diagrams [21].
Polarization-Dependent Measurements using polarized light on oriented samples provide information about transition moment directions, helping assign electronic transitions to specific symmetry species.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table: Key Research Reagents and Materials for Studying Selection Rules
| Reagent/Material | Function/Application | Example Use |
|---|---|---|
| Octahedral transition metal complexes (e.g., [Cr(NH₃)₆]³⁺, [CoF₆]³⁻) | Model systems for studying Laporte-forbidden d-d transitions | Demonstrating weak transition intensities in centrosymmetric environments |
| Tetrahedral transition metal complexes (e.g., [CoCl₄]²⁻, [MnO₄]⁻) | Model systems for studying relaxed selection rules | Comparing intense d-d transitions in non-centrosymmetric environments |
| Rare-earth doped materials (e.g., NaYF₄:Yb³⁺, Er³⁺) | Studying f-f transitions and upconversion processes | Investigating parity-forbidden transitions in asymmetric environments |
| Cryogenic solvents (e.g., liquid nitrogen, helium) | Temperature control for spectral resolution | Resolving vibrational fine structure in electronic spectra |
| Polarizers and optical components | Polarization-dependent spectroscopy | Determining transition moment directions and symmetries |
Visualization of Selection Rule Concepts
Relationship Between Selection Rules and Transition Intensity
Diagram: Selection Rule Decision Pathway and Transition Intensity
Vibronic Coupling Mechanism
Diagram: Vibronic Coupling Relaxing Laporte Rule
Selection rules, particularly the Laporte and spin selection rules, provide the fundamental framework for understanding electronic transitions in atoms and molecules. While these rules rigorously predict whether transitions are allowed or forbidden based on symmetry principles, real-world spectroscopic observations demonstrate that "forbidden" transitions regularly occur through relaxation mechanisms, most notably vibronic coupling.
The interplay between strict selection rules and their relaxation through vibronic coupling, spin-orbit coupling, and symmetry lowering creates the rich tapestry of spectroscopic phenomena observed in chemical systems. Understanding these principles enables researchers to interpret electronic spectra, design materials with tailored optical properties, and advance applications in fields ranging from solar energy conversion to biomedical imaging.
The continued study of selection rules and their manifestations remains essential for pushing the boundaries of spectroscopic science and its applications across chemistry, materials science, and related disciplines.
This technical guide explores the interpretation of fine structure in molecular spectra, focusing on the intricate coupling between vibrational and rotational energy transitions. Intended for researchers and drug development professionals, this whitepaper examines how fine structure analysis provides critical insights into molecular architecture and dynamics, framed within the broader context of electron energy level transitions spectroscopy. We present detailed experimental protocols, quantitative data relationships, and visualization tools to enable precise spectral interpretation in research applications, with particular emphasis on validating quantum chemical calculations through precision measurements.
Electronic spectroscopy relies on the quantized nature of energy states, where electrons absorb energy and transition from ground states to higher-energy excited states [22]. Without sufficient energy incentive, electrons will not transition to higher levels, but once excited, they subsequently relax to more stable states, releasing energy as photons detectable through spectroscopy [22]. Within this framework, vibrational and rotational fine structure provides the critical resolution needed to extract detailed molecular information.
The energy hierarchy of molecules follows a specific structure: embedded within electronic states (n=1,2,3...) are vibrational levels (v=1,2,3...), and within these exist rotational energy levels (J=1,2,3...) [22]. This nesting creates a complex energy landscape where transitions can involve changes in electronic, vibrational, and rotational states simultaneously. The ability to resolve this fine structure is paramount for advanced molecular analysis, particularly in pharmaceutical research where molecular structure directly correlates with biological activity.
Theoretical Foundation of Fine Structure
Quantum Mechanical Framework
In the quantum mechanical treatment of molecules, the total internal energy can be approximated as the sum of electronic, vibrational, and rotational components: [ E{\text{total}} = E{\text{electronic}} + E{\text{vibrational}} + E{\text{rotational}} ] For diatomic molecules, the vibrational energy levels are described by: [ G(v) = \omegae \left(v + \frac{1}{2}\right) - \omegae \chie \left(v + \frac{1}{2}\right)^2 ] where (v) is the vibrational quantum number, (\omegae) is the harmonic wavenumber, and (\chi_e) is the anharmonicity constant [23].
The rotational term values, to a first approximation, are given by: [ Fv(J) = Bv J(J+1) - D J^2 (J+1)^2 ] where (J) is the rotational quantum number, (B_v) is the rotational constant dependent on vibrational state, and (D) is the centrifugal distortion constant [23].
Selection Rules and Transition Patterns
The selection rules governing transitions between these energy states dictate the observed spectral patterns:
- Vibrational selection rules: For fundamental transitions, (\Delta v = \pm 1), though overtones with (\Delta v = \pm 2, \pm 3) etc. may occur with diminished intensity [23] [24].
- Rotational selection rules: For diatomic molecules, (\Delta J = \pm 1), giving rise to characteristic P- and R-branches [24].
- Electronic transition rules: Governed by symmetry considerations including spin, orbital, and symmetry rules [22].
These selection rules combine to produce the characteristic branch structure observed in high-resolution spectra, with the absence of a Q-branch ((\Delta J = 0)) in most heteronuclear diatomic molecules [23].
Rovibrational Coupling
Rotational-vibrational coupling represents a critical phenomenon where rotation and vibration frequencies interact, significantly affecting spectral appearance [25]. This coupling occurs because the rotational constant (Bv) depends on the vibrational quantum number due to the dependence of the moment of inertia on bond length: [ Bv = \frac{h}{8\pi^2 c Iv} ] where the moment of inertia (Iv = \mu dv^2) depends on the reduced mass (\mu) and the bond length (dv), which varies with vibrational state [23]. This relationship means that as a molecule vibrates, its bond length changes, affecting the moment of inertia and thus the rotational constant.
Table 1: Fundamental Energy Relationships in Rovibrational Spectroscopy
| Parameter | Symbol | Equation | Physical Significance |
|---|---|---|---|
| Vibrational Term | (G(v)) | (\omegae (v + \frac{1}{2}) - \omegae \chi_e (v + \frac{1}{2})^2) | Vibrational energy with anharmonic correction |
| Rotational Constant | (B_v) | (\frac{h}{8\pi^2 c I_v}) | Relationship between moment of inertia and rotational energy |
| Rotational Term | (F_v(J)) | (B_v J(J+1) - D J^2 (J+1)^2) | Rotational energy with centrifugal distortion |
| R-Branch Transition | (\nu_R) | (\nu_0 + 2B + (3B - B')J + (B - B')J^2) | Transitions with (\Delta J = +1) |
| P-Branch Transition | (\nu_P) | (\nu_0 - (B + B')J + (B - B')J^2) | Transitions with (\Delta J = -1) |
Experimental Methodologies
Precision Spectroscopy Techniques
Precision spectroscopic measurements, such as those conducted on the helium dimer (He₂), achieve remarkable resolution levels ((\Delta\nu/\nu = 2.5 \times 10^{-10})) with full resolution of rotational, spin-spin, and spin-rotational fine structures [26]. These experiments employ sophisticated laser systems and ultra-high vacuum environments to minimize broadening effects, enabling the detection of subtle quantum phenomena including tunneling predissociation through potential barriers [26].
For the (c^{3}\Sigma{g}^{+} \leftarrow a^{3}\Sigma{u}^{+}) electronic transition in He₂, researchers measure transitions to rotational levels located energetically above the dissociation limit, which decay by tunneling predissociation through a barrier in the potential energy surface [26]. This requires precise control of experimental conditions to observe these quasibound shape resonances.
Fourier-Transform Infrared (FTIR) Spectrometry
The NIST Quantitative Infrared Database employs FTIR spectrometry to measure absorption coefficient spectra of volatile organic compounds [27]. The fundamental equation governing these measurements is: [ It(\nu) = I0(\nu)10^{-acl} ] where (It) and (I0) are the transmitted and incident light intensities, (c) denotes the sample amount-of-substance concentration, and (l) is the path length [27]. Spectra are measured at multiple resolutions (from 2.00 cm⁻¹ to 0.125 cm⁻¹) using various apodization functions to optimize line shape accuracy [27].
Temperature Control and Solvent Effects
Temperature significantly affects spectral resolution of fine structure. Lower temperatures reduce thermal broadening and minimize population of excited vibrational states, allowing vibrational fine structure to emerge clearly [22]. Cryogenic techniques are often employed in high-resolution studies to maximize spectral detail. Similarly, solvent choice impacts spectral appearance through broadening mechanisms and shifts in energy levels, necessitating careful solvent selection for specific applications [22].
Data Interpretation and Analysis
Branch Structure Identification
Rotational fine structure manifests as distinct branches in vibrational spectra:
- R-branch: Corresponds to (\Delta J = +1) transitions, appearing at higher frequencies relative to the pure vibrational transition [23].
- P-branch: Corresponds to (\Delta J = -1) transitions, appearing at lower frequencies relative to the pure vibrational transition [23].
- Q-branch: Corresponds to (\Delta J = 0) transitions, which may be forbidden in some molecules, appearing at the fundamental vibrational frequency [23].
The method of combination differences provides a powerful technique for extracting rotational constants from complex spectra by analyzing energy differences between transitions sharing common upper or lower levels [23].
Molecular Constant Determination
The combination differences method allows separation of ground and excited state molecular constants. For the ground state: [ \Delta2^{\prime\prime} F(J) = \bar{\nu}(R(J-1)) - \bar{\nu}(P(J+1)) = (2B^{\prime\prime} - 3D^{\prime\prime})(2J+1) - D^{\prime\prime}(2J+1)^3 ] Similarly, for the excited state: [ \Delta2^{\prime} F(J) = \bar{\nu}(R(J)) - \bar{\nu}(P(J)) = (2B^{\prime} - 3D^{\prime})(2J+1) - D^{\prime}(2J+1)^3 ] These relationships enable determination of (B^{\prime\prime}) and (B^{\prime}), the rotational constants for ground and excited vibrational states, which in turn provide information about bond lengths in each state [23].
Table 2: Experimentally Derived Molecular Constants for He₂ Triplet States
| State | Vibrational Level | Rotational Constant B (cm⁻¹) | Centrifugal Distortion D (cm⁻¹) | Maximum N | Fine Structure Components |
|---|---|---|---|---|---|
| (a^{3}\Sigma_{u}^{+}) | v=0 | Derived from combination differences | Not specified | 9 | Full set for N up to 9 |
| (c^{3}\Sigma_{g}^{+}) | v=4 | Derived from combination differences | Not specified | 10 | Full set for N up to 10 |
| Transition | Precision | Branch Structure | Special Features | Theoretical Comparison | Dissociation Limit |
| (c^{3}\Sigma{g}^{+} \leftarrow a^{3}\Sigma{u}^{+}) | (2.5 \times 10^{-10}) | Full resolution of R and P branches | Predissociation broadening for N=10 | Parallel first-principles calculations | Above He(1¹S₀)+He(2³S₁) |
Advanced Analytical Techniques
Modern spectroscopic analysis employs sophisticated chemometric methods for data interpretation [28]:
- Signal Preprocessing: Baseline subtraction, derivative processing, normalization, and standard normal variate transformation enhance spectral quality [28].
- Component Analysis: Principal Component Analysis (PCA), Multivariate Curve Resolution (MCR), and Independent Component Analysis (ICA) resolve overlapping spectral features [28].
- Quantitative Methods: Partial Least Squares (PLS) regression, Principal Component Regression (PCR), and Multiple Linear Regression (MLR) enable quantitative determination of chemical parameters [28].
- Classification Methods: Soft Independent Modeling by Class Analogy (SIMCA), Support Vector Machines (SVMs), and Random Forest algorithms facilitate sample classification [28].
Case Study: Helium Dimer Fine Structure
The helium dimer serves as an exemplary system for studying fine structure, with four electrons making it amenable to first-principles quantum chemical calculations including nonadiabatic, relativistic, and quantum-electrodynamics corrections [26]. Precision measurements of the (c^{3}\Sigma{g}^{+} \leftarrow a^{3}\Sigma{u}^{+}) electronic transition at 2.5×10⁻¹⁰ precision provide critical tests for theoretical calculations [26].
In the (a^{3}\Sigma{u}^{+}) and (c^{3}\Sigma{g}^{+}) states of ⁴He₂, each rotational level with quantum number N splits into three fine-structure components with total angular momentum quantum number J = N, N±1 due to spin-spin and spin-rotation interactions [26]. The selection rules for electric dipole transitions in this system include ΔJ = 0, ±1 and ΔN = ±1, producing characteristic branch patterns [26].
A key finding involves the (c^{3}\Sigma_{g}^{+}(v=4, N=10)) levels, which show pronounced broadening due to tunneling predissociation through a barrier in the potential energy curve [26]. This phenomenon provides quantitative information about potential energy surfaces and decay dynamics, demonstrating how fine structure analysis reveals detailed molecular dynamics information beyond basic structural parameters.
Computational Modeling and Theoretical Validation
Advanced theoretical treatments now enable precise calculation of molecular fine structure. For He₂, quantum-chemical calculations include nonadiabatic effects, relativistic corrections, and quantum-electrodynamics contributions, providing predictions testable against experimental measurements [26]. The close agreement between theory and experiment for helium dimer fine structure intervals validates both the computational methods and the experimental approaches [26].
For complex molecules, computational spectroscopy combines quantum chemistry calculations with spectral simulation to predict and interpret fine structure. These approaches leverage potential energy surface calculations, dipole moment functions, and variational methods to simulate rovibrational spectra for comparison with experimental data.
Applications in Pharmaceutical Research
In drug development, vibrational and rotational fine structure analysis provides critical insights into molecular structure and interactions:
- Conformational Analysis: Fine structure reveals subtle conformational changes affecting drug-receptor interactions.
- Hydration State Determination: Spectral shifts and broadening indicate solvation effects relevant to bioavailability.
- Polymorph Identification: Distinct fine structure patterns differentiate crystalline forms with implications for drug stability and dissolution.
- Binding Constant Determination: Spectral changes upon ligand binding quantify interaction strengths.
The emergence of vibrational fine structure at reduced temperatures provides enhanced resolution for characterizing pharmaceutical compounds, enabling more precise structural assignments [22].
Visualization of Spectral Transitions
Essential Research Reagents and Materials
Table 3: Essential Research Reagent Solutions for Precision Spectroscopy
| Reagent/Equipment | Function/Purpose | Technical Specifications | Application Example |
|---|---|---|---|
| Primary Gas Standards | Reference for concentration calibration | Certified purity, verified amount-of-substance fraction | NIST quantitative IR database development [27] |
| Cryogenic Cooling Systems | Temperature control for resolution enhancement | Capable of sub-10K operation | Helium dimer fine structure studies [26] |
| FTIR Spectrometer | High-resolution spectral acquisition | Multiple apodization functions, 0.125 cm⁻¹ resolution | Volatile organic compound analysis [27] |
| Ultra-High Vacuum Chambers | Elimination of broadening effects | Pressure < 10⁻⁹ mbar | Precision measurements of triplet states [26] |
| Tunable Laser Systems | Precise excitation source | Narrow linewidth, frequency stabilization | Resolving spin-rotation interactions [26] |
| Reference Spectral Databases | Data validation and compound identification | Curated collections with uncertainty estimates | SDBS, NIST WebBook, HITRAN [29] |
Vibrational and rotational fine structure analysis represents a powerful methodology for extracting detailed molecular information from spectral data. Through advanced experimental techniques including precision laser spectroscopy and FTIR spectrometry, combined with sophisticated theoretical models and computational chemistry, researchers can resolve intricate details of molecular structure, dynamics, and interactions. The continuing refinement of these approaches, particularly through synergistic experimental-theoretical collaborations as demonstrated in helium dimer research, promises enhanced capabilities for molecular analysis across chemical, pharmaceutical, and materials sciences.
The transition of electrons between energy levels forms the foundation of elemental spectrochemical analysis, enabling techniques in both the optical and X-ray regions of the electromagnetic spectrum [30]. While both optical and X-ray transitions originate from electron energy changes within atoms, they operate on vastly different energy scales and involve fundamentally distinct electron shells [30]. Optical transitions typically involve outer-shell electrons and produce electromagnetic radiation in the ultraviolet, visible, and infrared regions with wavelengths from approximately 100 to 1000 nm [30]. In contrast, X-ray transitions result from the excitation of inner-shell electrons, producing electromagnetic radiation with much shorter wavelengths from about 0.01 to 10 nm [30]. This fundamental difference in the electron shells involved—outer valence electrons for optical transitions versus inner core electrons for X-ray transitions—creates an energy separation of approximately three orders of magnitude between these processes [30].
The energy disparity arises directly from the significantly higher binding energies of inner-shell electrons compared to those in outer shells [30] [31]. For example, in magnesium (Z = 12), the binding energy of K-shell electrons is about three orders of magnitude greater than the ionization-excitation energies relevant to optical emission spectroscopy [30]. This energy differential fundamentally shapes the required excitation sources, instrumentation, and applications for each spectroscopic method, making each technique uniquely suited for different analytical challenges in scientific research and industrial applications, including pharmaceutical development [32] [33].
Theoretical Framework of Electron Transitions
Atomic Energy Level Structure
In atomic physics, electron energy levels are grouped based on the principal quantum number n, with these groups differing from each other by approximately a factor of 10 in binding energy [30]. The standard electron energy level designations include the principal quantum number "n," the orbital angular momentum quantum number "l," and the magnetic quantum number "m" [30]. In X-ray spectroscopy, the innermost electron shell is called the K-shell (n=1); the next farthest from the nucleus is the L-shell (n=2), followed by the M-shell (n=3), and so forth [30]. This nomenclature provides a systematic framework for understanding the origin of characteristic X-ray spectral lines.
For optical transitions, the energy level diagram is structured differently, with the ground state of the neutral atom typically considered the zero reference level for various excited states and ionization levels [30]. This conceptual difference in energy level referencing highlights the challenge in creating a unified diagram that accurately represents both X-ray and optical transitions simultaneously. When such a combined diagram is attempted, mathematical irregularities inevitably appear due to the vastly different energy scales involved [30].
Selection Rules and Transition Probabilities
Atomic transitions must obey specific selection rules that follow from principles of quantum mechanics and symmetry [31]. These rules classify transitions as either allowed or forbidden, with forbidden transitions occurring with very low probability [31]. For hydrogen-like atoms, atomic transitions involving electromagnetic interactions (emission and absorption of photons) obey the selection rule Δl = ±1, where l is associated with the magnitude of orbital angular momentum [31]. For multi-electron atoms, similar rules apply, though the presence of multiple electrons introduces additional complexity due to electron-electron interactions and internal magnetic interactions (spin-orbit and spin-spin couplings) [31].
The probability of different transition types varies significantly. In X-ray fluorescence, the process occurs when an outer-shell electron "drops down" to fill a void created in an inner shell, with a certain probability that an X-ray characteristic of that atom will be emitted [30]. For optical emissions, the excitation energy must provide the necessary energy to either move an electron of the neutral atom from its ground state to an excited state, remove an electron from the atom (ionization), or excite an electron of the ion into an excited state of that ion [30]. The subsequent return of excited electrons to lower energy levels releases energy as optical radiation.
Transition Processes Visualization
Quantitative Energy Comparison
The energy disparity between optical and X-ray transitions represents the most significant differentiating factor between these processes. The table below summarizes key quantitative differences:
Table 1: Energy Scale Comparison Between Optical and X-ray Transitions
| Parameter | Optical Transitions | X-ray Transitions | Ratio (X-ray/Optical) |
|---|---|---|---|
| Typical Energy Range | 1-10 eV [30] | 100 eV - 100 keV [30] [33] | ~100-10,000x |
| Wavelength Range | 100-1000 nm (UV-Vis-IR) [30] | 0.01-10 nm [30] | ~0.00001-0.1x |
| Electron Shells Involved | Outer valence shells [30] | Inner core shells (K, L, M) [30] | N/A |
| Binding Energy of Target Electrons | ~1-20 eV [30] | ~100 eV - 100 keV [30] [33] | ~100-10,000x |
| Excitation Sources | Arc/spark, laser, plasma [30] | X-ray tube, synchrotron [33] | N/A |
| Ionization Potential | ~5-25 eV [30] | N/A (ejection, not ionization) | N/A |
The substantial energy difference profoundly impacts the physical processes involved. For X-ray transitions to occur, the excitation energy must equal or exceed the binding energy required to remove an electron from its inner shell, following the relationship E ≥ EB, where EB represents the binding energy [30]. In the X-ray fluorescence process, the binding energy of innermost electrons represents the critical quantity [30]. For instance, if the energy of incoming excitation is insufficient to eject a K-shell electron, no characteristic X-rays are produced [30].
For optical emission spectroscopy, the process differs significantly. Here, excitation energy must provide the necessary energy for: (1) moving an electron of the neutral atom from its ground state to an excited state; (2) removing an electron from the atom (ionization); or (3) exciting an electron of the ion into an excited state of that ion [30]. While the relationship E ≥ EB still applies for ionization, a better formulation for excitation might be E ≥ Ee, where Ee represents the appropriate atomic or ionic excitation energy [30].
Table 2: Characteristic Transition Energies for Selected Elements
| Element | Optical Transition (eV) | X-ray Kα Energy (eV) | K-shell Binding Energy (eV) | Ionization Potential (eV) |
|---|---|---|---|---|
| Magnesium (Z=12) | ~4.3 (Mg I 285.21 nm) [30] | ~1250 [30] | ~1300 [30] | 7.6 [30] |
| Carbon (Z=6) | N/A | ~280 [34] | ~284 [33] | 11.3 |
| Iron (Z=26) | N/A | ~6400-6900 [35] [34] | ~7100 [35] | 7.9 |
| Manganese (Z=25) | N/A | ~5900-6490 [36] [35] | ~6540 [35] | 7.4 |
Spectroscopic Techniques and Methodologies
X-ray Absorption and Emission Spectroscopy
X-ray Absorption Spectroscopy (XAS) measures changes in the absorption coefficient (μ) of a material as a function of incident photon energy, providing information about the density of unoccupied electronic states and the local atomic structure around the absorbing atom [33]. Each element has a characteristic absorption edge representing a sharp increase in absorption when photon energy reaches the binding energy of a core-level electron [33]. This element-specific edge makes XAS highly selective, allowing targeted study of chosen elements through appropriate tuning of excitation energy [33].
X-ray Emission Spectroscopy (XES) involves analyzing photons emitted when core holes created during X-ray absorption are filled by electrons from higher shells [33] [35]. The recombination process is accompanied by emission of radiation with energy equal to the difference between the two levels, characteristic of a given element [33]. For transition metals, the Kβ emission spectrum contains detailed information about spin and oxidation state, with the Kβ1,3 and Kβ' regions split through a 3p-3d exchange interaction [35]. Weaker features in the Kβ2,5 and Kβ″ regions provide information about ligand-atom type, distance, and orientation [35].
Optical Emission Spectroscopy Methodologies
Optical emission spectroscopy techniques involve exciting outer-shell electrons to higher energy states using sources such as arcs, sparks, lasers, or plasmas [30]. When these excited electrons return to lower energy levels, they emit photons with energies corresponding to the difference between the excited and ground states [30] [31]. These transitions appear as sharp spectral lines after passing through a spectrometer [31]. In modern nomenclature, optical spectral lines are designated with Roman numerals: I for the neutral atom, II for the first ionized state, and so forth [30]. For example, the prominent magnesium lines are written as Mg I 285.21 nm and Mg II 279.55 nm [30].
Experimental Workflow
Research Reagent Solutions and Essential Materials
Successful implementation of spectroscopic techniques requires specific instrumentation and materials. The following table outlines essential components for both X-ray and optical spectroscopy:
Table 3: Essential Research Reagents and Materials for Spectroscopy
| Category | Specific Items/Components | Function/Purpose | Technique Application |
|---|---|---|---|
| Excitation Sources | Synchrotron radiation, X-ray tubes [33] | Provides high-energy photons for core electron ejection | XAS, XES, XRF |
| Arc/spark sources, lasers, plasma sources [30] | Excites valence electrons to higher energy states | OES | |
| Detection Systems | Energy-dispersive detectors, wavelength-dispersive spectrometers [33] [35] | Measures energy/wavelength of emitted X-rays | XES, XRF |
| Grating spectrometers, photomultiplier tubes [30] | Disperses and detects optical photons | OES | |
| Sample Preparation | Hydraulic presses, pellet dies | Prepens powdered samples as pellets for analysis | XAS, XES |
| Liquid sample cells, thin films | Presents liquid samples in appropriate geometry | XAS (fluorescence mode) | |
| Reference Materials | Pure elemental foils (Cu, Fe) [35] | Energy calibration and spectrometer alignment | XAS, XES |
| Certified standard reference materials | Quantitative calibration and method validation | OES, XRF | |
| Specialized Components | Multi-crystal analyzers [35] | High-resolution X-ray fluorescence detection | XES |
| Ionization chambers [33] | Measures incident and transmitted X-ray intensity | XAS (transmission mode) |
Pharmaceutical and Biomedical Applications
Drug Development and Analysis
X-ray absorption and emission spectroscopy techniques enable precise analysis of electronic structure and local atomic environment in pharmaceutical compounds [33]. These methods support studies on catalytic mechanisms, redox processes, and metal speciation in drug molecules [33]. A key advantage is their element selectivity, allowing analysis of specific elements without matrix interference [33]. Their high sensitivity to chemical state and coordination enables determination of oxidation states, electronic configuration, and local geometry, which is particularly valuable for metallodrugs and metal-containing pharmaceutical compounds [33].
These techniques are applicable to solids, liquids, and gases without special sample preparation, making them valuable for analyzing various drug formulations [33]. Modern XAS and XES studies are typically performed using synchrotron radiation, which provides intense, monochromatic X-ray sources and allows advanced in situ and operando experiments [33]. Sub-techniques such as XANES (X-ray absorption near-edge structure), EXAFS (Extended X-ray Absorption Fine Structure), and RIXS (resonant inelastic X-ray scattering) offer enhanced insights into oxidation states, local structure, and electronic excitations relevant to drug behavior and stability [33].
Biomedical Research Applications
In biomedical research, X-ray spectroscopy methods have been applied to characterize metalloproteins and enzyme active sites [35]. For example, Kβ X-ray fluorescence spectroscopy has been used to characterize the oxidation states of the manganese cluster in photosystem II, the protein complex responsible for photosynthetic splitting of water and oxygen release [35]. Studies of the Mn cluster in the Kok cycle have helped resolve questions about whether oxidation steps are metal-centered or ligand-centered, with important implications for understanding the mechanism of photosynthetic water splitting [35].
Another biocatalytic application involves studying manganese oxidation by bacteria, such as Bacillus SG-1 spores [35]. Kβ1,3 X-ray fluorescence spectroscopy has provided insights into whether biological manganese oxidation proceeds through a one-electron or two-electron process, helping interpret anomalous concentrations of manganese oxides in geological rock strata and potential biosignatures for studies of early life [35].
Synergistic Diagnostic and Therapeutic Applications
The complementary strengths of X-ray and optical technologies are being exploited in emerging biomedical applications [32]. X-rays provide deep tissue penetration, while optical interactions offer molecular specificity [32]. An emerging trend is the integration of nanoparticles to serve as molecular intermediates or energy transducers for imaging and therapy [32]. These nanoscale designs are impacted by choices of optical interaction mechanism, such as scintillation or Cherenkov light [32]. The enhancement of optical molecular sensing or sensitization of tissue using X-rays as the energy source represents an important emerging field combining X-ray tissue penetration in radiation oncology with the molecular specificity and packaging of optical probes or molecular localization [32].
The comparative analysis of optical versus X-ray transitions reveals a fundamental energy scale difference of approximately three orders of magnitude, stemming from the distinct electron shells involved in each process. Optical transitions, involving valence electrons with binding energies of ~1-20 eV, provide information about molecular orbitals and chemical bonding, while X-ray transitions, involving core electrons with binding energies from hundreds to thousands of eV, offer element-specific information about local atomic structure and oxidation states. This energy differential dictates the required instrumentation, with optical spectroscopy employing arc/spark sources and grating spectrometers, while X-ray spectroscopy requires synchrotron radiation or X-ray tubes with energy-dispersive detectors. Despite their differences, both techniques provide complementary information that continues to advance fields ranging from fundamental physics to pharmaceutical development and biomedical research. The ongoing integration of these technologies, particularly through nanoparticle intermediaries, promises new capabilities in diagnostic and therapeutic applications that leverage the unique strengths of both energy regimes.
Spectroscopy in Action: Analytical Techniques for Drug Development and Diagnostics
Electronic Spectroscopy for Molecular Characterization and Binding Studies
Electronic spectroscopy probes the quantized energy states of molecules, providing critical insights into their electronic structure, identity, and interactions. This family of techniques relies on the fundamental principle that electrons within molecules can be excited from a ground state to a higher energy excited state by absorbing photons [22]. The energy required for these electron transitions is characteristic of a molecule's specific structure and environment. When an electron absorbs a photon with energy (hν) matching the gap between its current orbital and an available higher-energy orbital, it undergoes a transition, recorded as an absorption band in a spectrum [22]. The process is cyclical; after excitation, the electron relaxes back to its ground state, often releasing energy as photons through fluorescence [9].
The significance of electronic spectroscopy in molecular characterization lies in its ability to serve as a fingerprinting tool. The specific wavelengths of light a molecule absorbs, and the intensity of that absorption, are dictated by the energy differences between molecular orbitals and the probability of the transitions between them [22]. These spectral features are sensitive to the molecular framework, substituents, and the solvent environment, allowing researchers to identify functional groups, probe conjugation, and study molecular interactions [22] [37]. Furthermore, within the broader context of electron energy level transition spectroscopy, electronic absorption spectroscopy is a cornerstone technique. It complements other methods like photoelectron spectroscopy, which directly probes orbital energies by measuring the kinetic energy of ejected electrons, and inelastic electron tunneling spectroscopy (IETS), which probes vibrational modes via electron-vibration interactions in molecular junctions [38] [39].
Theoretical Foundations of Electronic Transitions
The Jablonski Diagram and Energy Transfer Processes
The processes of absorption and emission are most commonly visualized using a Jablonski diagram, which maps the electronic and vibrational energy levels of a molecule and the transitions between them [9]. This diagram is indispensable for understanding the pathways of energy flow following photon absorption.
The following diagram illustrates the key stages of the fluorescence process and competing pathways:
The fluorescence process is a three-stage cycle [9]:
- Excitation: A photon of energy
hν_EXis absorbed, promoting the fluorophore to an excited electronic singlet state (S₁'). - Excited-State Lifetime: The fluorophore exists in this state for 1–10 nanoseconds, undergoing conformational changes and interactions with its environment. This results in a loss of energy, yielding a relaxed singlet excited state (
S₁). - Emission: A photon of lower energy
hν_EMis emitted as the fluorophore returns to its ground state (S₀). The energy difference betweenhν_EXandhν_EMis the Stokes shift, fundamental for isolating emission signals from excitation light [9].
Competing pathways like intersystem crossing to a triplet state (T₁) and collisional quenching can depopulate S₁ without emission, reducing the fluorescence quantum yield [9].
Selection Rules and Spectral Interpretation
Not all possible electron transitions are equally probable. Selection rules, derived from quantum mechanics, govern the allowed transitions, which appear as intense bands in a spectrum [22].
- Laporte Rule: For centrosymmetric molecules, transitions between orbitals of the same parity (e.g., g→g or u→u) are Laporte-forbidden. Transitions between a gerade (g) and ungerade (u) orbital (g→u) are allowed. This rule makes d-d transitions in symmetrical transition metal complexes formally forbidden, though they gain weak intensity through vibronic coupling [22] [40].
- Spin Rule: Transitions that involve a change in the electron's spin state (e.g., singlet→triplet) are spin-forbidden. They occur with very low probability compared to spin-allowed transitions (singlet→singlet) [22].
The interpretation of an absorption spectrum involves assigning the observed bands to specific electronic transitions. Table 1 summarizes common types of transitions and their spectral characteristics.
Table 1: Common Electronic Transitions and Their Spectral Signatures
| Transition Type | Typical Energy Range | Molar Extinction Coefficient (ε) | Example Compounds |
|---|---|---|---|
| σ → σ* | High UV | Very High | Alkanes |
| n → π* | UV-Vis | Low (10-100 L mol⁻¹ cm⁻¹) | Carbonyls |
| π → π* | UV-Vis | High (10,000-250,000 L mol⁻¹ cm⁻¹) | Alkenes, Aromatics |
| Charge Transfer | UV-Vis-NIR | Very High | Metal Complexes |
| d-d* | Vis-NIR | Low (10-500 L mol⁻¹ cm⁻¹) | Transition Metal Complexes [40] |
*Formally Laporte-forbidden, so intensity is low.
The presence of vibrational fine structure within a broader electronic absorption band, often resolved at low temperatures, provides further information about the vibrational energy levels of the molecule in its ground and excited states [22].
Experimental Methodologies and Protocols
Core Instrumentation and Workflow
Electronic absorption spectroscopy instruments, often UV-Vis spectrophotometers, are designed to measure the absorption of light as a function of wavelength. Fluorescence instruments, or spectrofluorometers, have a similar core design but are optimized for detecting emitted light [9].
The general workflow for an electronic absorption experiment involves several key stages, from sample preparation to data analysis, as illustrated below:
Detailed Experimental Protocol: DNA Binding Studies
UV-Vis spectroscopy is a well-established method for quantifying the interaction of small molecules with biomacromolecules like DNA [37]. The following protocol is adapted from studies of imidazolidine derivatives [37].
Objective: To determine the binding constant of a small molecule (e.g., an imidazolidine derivative) with DNA.
Materials and Reagents:
- Tris-HCl or phosphate buffer (e.g., 5 mM NaH₂PO₄, 50 mM NaCl, pH 7.0): Provides a stable, physiologically relevant ionic environment [37].
- Calf Thymus DNA (CT-DNA) or synthetic oligonucleotide: The source of double-stranded DNA.
- Test compound solution: The small molecule under investigation, dissolved in a compatible solvent (e.g., DMSO, with final concentration <1% v/v).
- UV-Vis spectrophotometer with matched quartz cuvettes (typically 1 cm pathlength).
- Temperature-controlled cuvette holder.
Procedure:
- DNA Stock Solution Preparation: Dissolve CT-DNA in buffer. Verify the purity by ensuring the ratio of absorbance at 260 nm to that at 280 nm is between 1.8 and 1.9. Determine the concentration using the molar extinction coefficient at 260 nm (ε ≈ 6600 M⁻¹ cm⁻¹ per base pair).
- Sample Preparation: Prepare a fixed concentration of the test compound in a cuvette. This concentration should be within the linear range of the Beer-Lambert law for the compound.
- Titration Experiment: Record the baseline-corrected absorption spectrum of the test compound alone. Then, add small, incremental volumes of the DNA stock solution to the cuvette. Mix thoroughly and allow the solution to equilibrate for a few minutes before recording the spectrum after each addition.
- Data Collection: Continue the titration until no further changes in the absorption spectrum are observed, indicating binding saturation.
Data Analysis: Monitor changes in the absorption spectrum of the test compound upon each DNA addition. These changes can include:
- Hypochromism: A decrease in absorption intensity.
- Hyperchromism: An increase in absorption intensity.
- Red- or Blue-shift (Bathochromism or Hypsochromism): A shift of the absorption peak to longer or shorter wavelengths, respectively.
The binding constant (Kb) can be determined by applying the following equation, which models the interaction:
Analysis requires plotting the change in absorbance (ΔA) against the concentration of DNA. The data can be fit using non-linear regression or a linear transformation, such as a Scatchard or Benesi-Hildebrand plot, to calculate Kb [37]. A greater Kb value indicates a stronger binding affinity.
Applications in Molecular Characterization and Binding Studies
Quantitative Binding Analysis in Drug Discovery
Electronic spectroscopy provides quantitative and qualitative data on molecular interactions critical to pharmaceutical development. In a study on imidazolidine derivatives, UV-Vis titration and cyclic voltammetry were used to determine DNA binding constants at physiological pH (7.4), revealing a significant range of affinities [37]. The results, summarized in Table 2, demonstrate how spectroscopy can rank drug candidates.
Table 2: DNA Binding Constants of Imidazolidine Derivatives Determined by Electronic Absorption Spectroscopy [37]
| Compound Abbreviation | Full Name | Binding Constant (K_b) at pH 7.4 (M⁻¹) | Inference |
|---|---|---|---|
| NBI | 5-benzylideneimidazolidine-2,4-dione | 6.40 × 10⁶ | Strongest binder; preferred candidate |
| HBI | 5-(2-hydroxybenzylidene)imidazolidine-2,4-dione | 1.77 × 10⁵ | Moderate binder |
| MBI | 5-(4-methoxybenzylidene)imidazolidine-2,4-dione | 2.06 × 10⁴ | Weak binder |
| DBI | 5-(3,4-dimethoxybenzylidene)imidazolidine-2,4-dione | 1.01 × 10⁴ | Weakest binder |
Monitoring Conformational and Stability Changes
Electronic absorption spectroscopy is a powerful tool for probing the stability and structural changes of biomolecules. A classic application is determining the melting temperature (Tm) of DNA.
- Protocol: The absorbance of a DNA solution at 260 nm is monitored as the temperature is gradually increased (e.g., 0.5°C/min). As the temperature rises, the double-stranded helix denatures into single strands. This unstacking of the bases leads to a ~40% increase in absorbance (hyperchromic effect). The melting temperature (Tm*) is defined as the temperature at the midpoint of this sigmoidal transition curve, where the rate of absorbance change is maximum [40].
- Interpretation: The Tm value provides insights into the stability of the DNA duplex. Longer duplexes and those with a higher GC content (three hydrogen bonds per base pair) have higher Tm than shorter or AT-rich duplexes [40]. Changes in Tm upon binding of a small molecule can also indicate the mode of interaction (e.g., intercalation often stabilizes the duplex and increases Tm).
Characterization of Metal Complexes and Nanoparticles
The technique is indispensable in inorganic chemistry and nanomaterial science. For transition metal complexes, the color arises from d-d transitions or charge-transfer transitions [40]. The number, energy, and intensity of absorption bands in the visible region can be used to deduce the geometry and oxidation state of the metal center. Similarly, the surface functionalization of nanoparticles and their subsequent interactions with biomolecules can be monitored and characterized using electronic spectroscopy and complementary techniques like Surface Plasmon Resonance (SPR) [41].
Advanced Techniques and Future Directions
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful experimentation in electronic spectroscopy requires careful selection of reagents and materials.
Table 3: Key Research Reagent Solutions for Electronic Spectroscopy Experiments
| Reagent/Material | Function | Example Use Case |
|---|---|---|
| High-Purity Buffers (e.g., Tris-HCl, Phosphate) | Maintain stable pH and ionic strength, mimicking physiological conditions. | DNA binding studies [37], protein folding analysis. |
| DNA Stock Solutions (e.g., Calf Thymus DNA) | The macromolecular target for binding studies. | Determining binding constants and mode of interaction of small molecules [37]. |
| Fluorescent Dyes/Probes | Target-specific labels for sensitive detection. | Tracking biomolecules in cells, FRET-based interaction studies [9]. |
| Reference Standards (e.g., fluorescent microspheres, quinine sulfate) | Calibrate instrument performance and enable quantitative comparisons. | Ensuring data reproducibility across different instruments and time points [9]. |
| Nanoparticle Suspensions | Functional materials for drug delivery and sensing. | Characterizing surface conjugation and stability using SPR and UV-Vis [41]. |
Emerging Trends and Complementary Spectroscopies
The field of molecular spectroscopy is continuously advancing. A significant trend is the push of traditionally low-temperature techniques toward room-temperature operation. For instance, Inelastic Electron Tunneling Spectroscopy (IETS), a powerful vibrational spectroscopy for molecular junctions, has recently been demonstrated at temperatures up to ~400 K, opening avenues for practical device characterization under ambient conditions [38].
The integration of machine learning for the analysis of complex spectral data, such as IETS, is another powerful advance, enabling automated interpretation and prediction of spectra [38].
Furthermore, electronic absorption spectroscopy is increasingly used in conjunction with other techniques in a multimodal approach. Surface Plasmon Resonance (SPR) is a key complementary technology that provides real-time, label-free data on binding kinetics (association/dissociation rates) that UV-Vis absorption cannot easily measure [41]. SPR is extensively used in nanoparticle research to monitor synthesis, functionalization, and interactions with biomolecules [41].
Near-Infrared (NIR) Spectroscopy in Non-Destructive Pharmaceutical Analysis
Near-infrared (NIR) spectroscopy has emerged as a cornerstone analytical technique within the pharmaceutical industry, distinguished by its rapid, non-destructive nature and minimal requirement for sample preparation. This spectroscopic method utilizes the near-infrared region of the electromagnetic spectrum, spanning from approximately 780 to 2500 nanometers (12,820 to 3,959 cm⁻¹), to probe the chemical and physical properties of pharmaceutical materials [42] [43]. The technique's ability to provide multi-constituent analysis of virtually any matrix has cemented its role from raw material identification to final product quality control [42]. The growing pharmaceutical interest is a direct result of its major advantages: the possibility of separating the sample measurement position and spectrometer by use of fiber optic probes, and the prediction of chemical and physical sample parameters from a single spectrum [42]. Framed within the broader context of electron energy level transitions, NIR spectroscopy operates on principles distinct from UV-Vis spectroscopy. While UV-Vis involves the promotion of electrons to higher energy orbitals, the energy of NIR photons is sufficiently lower that electron promotions do not result from it [44]. Instead, NIR spectroscopy probes the vibrational states of molecules through overtones and combination bands of fundamental molecular vibrations, primarily those of C-H, O-H, and N-H functional groups [42] [43] [45].
Theoretical Foundation: Bridging Electron Transitions and Molecular Vibrations
The Electronic-Vibrational Bridge in NIR Spectroscopy
To fully appreciate NIR spectroscopy's analytical power, one must understand its position in the electromagnetic spectrum and its effect on matter. The NIR region resides between the higher-energy visible spectrum (where electronic transitions occur) and the lower-energy mid-infrared region (where fundamental molecular vibrations occur) [45]. This intermediary position dictates its unique interaction with molecules. The energy of NIR photons is insufficient to cause electronic transitions but is ideal for exciting molecules to higher vibrational states. These are not the fundamental vibrations measured in mid-IR spectroscopy, but rather overtones (transitions from the ground state to excitation levels above the first state) and combination bands (simultaneous excitation of two different vibrations) [43] [45]. These transitions are 10–100 times weaker in intensity than their fundamental mid-IR counterparts, which allows NIR radiation to penetrate much further into a sample, enabling the analysis of bulk material [43]. This theoretical foundation explains why NIR spectroscopy is exceptionally suited for the analysis of intact pharmaceutical dosage forms, as it probes the entire sample volume rather than just the surface.
Molecular Information Content in NIR Spectra
The most prominent absorption bands in the NIR region arise from the overtones and combinations of fundamental vibrations of –CH, –NH, –OH (and –SH) functional groups [42]. Because these functional groups are ubiquitous in active pharmaceutical ingredients (APIs) and excipients, NIR spectra contain a wealth of chemical information. However, the resulting spectra are typically composed of broad, overlapping absorption bands, making them visually complex and difficult to interpret with traditional univariate methods [42] [43]. To extract meaningful chemical and physical information, advanced chemometric (multivariate) data processing techniques are required. These mathematical and statistical methods, including principal component analysis (PCA) and partial least squares (PLS) regression, are essential for relating spectral features to analyte properties, thereby enabling both qualitative identification and quantitative analysis [42] [43].
Key Pharmaceutical Applications and Quantitative Data
The applications of NIR spectroscopy in the pharmaceutical industry are extensive and cover the entire product lifecycle, from raw material testing to manufacturing process monitoring and final product release. Its non-destructive nature is particularly valuable for maintaining product integrity and enabling real-time decisions.
Table 1: Quantitative Applications of NIR Spectroscopy in Pharmaceutical Analysis
| Application Category | Specific Measured Parameter | Reported Performance/Accuracy | Reference Technique |
|---|---|---|---|
| Raw Material Identification | Identity of APIs and excipients | High identification accuracy | Compendial methods [42] |
| Solid Dosage Form Analysis | API content in intact tablets | Quantitative analysis from single spectrum | HPLC [42] |
| Tablet hardness | Non-destructive prediction | Destructive hardness testing [42] | |
| Physical Form Characterization | Polymorph composition in powder mixtures | Low-level determination possible | XRPD [42] |
| Crystallinity | Quantitative analysis demonstrated | DSC, XRPD [42] | |
| Process Monitoring | Blend uniformity in continuous manufacturing | Monitors Critical Quality Attributes every few seconds | HPLC grab sampling [46] |
| Coating thickness and moisture content | Cuts batch-failure rates by double-digit percentages | Off-line lab testing [46] |
Qualitative and Quantitative Analysis
Qualitative applications are primarily focused on the identification and verification of raw materials, including APIs and excipients [42] [47]. The technique's speed and minimal sample preparation allow for high-throughput testing in a Good Manufacturing Practice (GMP) environment, significantly improving logistics and reducing the risk of cross-contamination. For quantitative analysis, NIR spectroscopy can determine both chemical parameters, such as the content of an API in an intact tablet, and physical parameters, such as polymorphic form, crystallinity, and moisture content [42]. The ability to analyze intact dosage forms without dissolution or destruction is a paramount advantage, enabling 100% product screening if desired. The quantification of water content is a particularly strong application, as the -OH groups of water produce distinct and sensitive spectral signatures in the NIR region [42].
Process Analytical Technology (PAT) and Real-Time Release
A transformative application of NIR spectroscopy is its role in Process Analytical Technology (PAT) for real-time process monitoring and control [47] [46]. Guided by regulatory frameworks encouraging quality-by-design, the pharmaceutical industry increasingly implements NIR probes directly into manufacturing equipment. This allows for continuous, non-destructive monitoring of Critical Quality Attributes (CQAs) during unit operations such as granulation, drying, blending, and tablet coating [47] [46]. For example, in-line NIR probes can monitor blend homogeneity in real-time, ensuring a uniform mixture before tableting and eliminating the need for time-consuming and statistically limited thief sampling. This real-time data facilitates the implementation of a real-time release testing (RTRT) strategy, where a product can be released based on process data meeting all predefined specifications, without the need for end-product testing [47]. This approach enhances efficiency, reduces production cycle times, and ensures a more consistent product quality.
Experimental Protocols and Methodologies
Implementing a robust NIR analytical method requires careful attention to experimental design, from instrument selection to model validation. The following protocols outline standard methodologies for key pharmaceutical applications.
Protocol for Raw Material Identification
Objective: To rapidly and non-destructively identify incoming raw materials (APIs, excipients) against a validated spectral library.
Materials and Equipment:
- NIR spectrometer (benchtop, portable, or handheld) with a reflectance probe or accessory.
- Representative samples of the raw material for library building and validation.
- Computer with chemometric software for spectral library creation and comparison.
Procedure:
- Library Development: Collect NIR spectra (e.g., 100 scans at 8 cm⁻¹ resolution) from multiple batches of each certified raw material. Ensure spectral acquisition covers the entire representative sample.
- Spectral Pre-processing: Apply standard pre-processing techniques to the library spectra, such as Standard Normal Variate (SNV) or Multiplicative Scatter Correction (MSC) to reduce physical light scattering effects, and first or second derivatives (e.g., Savitzky-Golay) to enhance spectral features and remove baseline offsets [42].
- Model Creation: Use a chemometric method like Principal Component Analysis (PCA) to create a model that defines the acceptable spectral variation for each material.
- Validation: Challenge the model with validation samples (known materials not used in the library) to establish identification confidence limits (e.g., Mahalanobis distance).
- Routine Testing: For an unknown sample, collect its spectrum, apply the same pre-processing, and compare it to the spectral library. The sample is positively identified if it falls within the pre-defined confidence limits of a library material and does not match any other.
Protocol for Quantitative Analysis of API in Intact Tablets
Objective: To determine the content of the Active Pharmaceutical Ingredient (API) in an intact tablet without destruction.
Materials and Equipment:
- NIR spectrometer equipped with a reflectance accessory for solids.
- A calibration set of tablets with known API concentrations, as determined by a reference method (e.g., HPLC).
- Validation set of tablets with known API concentrations.
Procedure:
- Calibration Set Selection: Assemble a set of tablets that encompasses the expected manufacturing variation in API concentration and physical properties (e.g., hardness, density).
- Reference Method Analysis: Determine the exact API content of each calibration tablet using the validated reference method (e.g., HPLC).
- Spectral Acquisition: Collect NIR spectra from all tablets in the calibration set without altering them.
- Chemometric Model Development: Use a quantitative algorithm like Partial Least Squares (PLS) regression to correlate the spectral data (X-matrix) with the reference API values (Y-matrix). The software identifies latent variables that best describe the covariance between the spectra and the concentration.
- Model Validation: Use the independent validation set of tablets to assess the model's predictive performance. Key figures of merit include Root Mean Square Error of Prediction (RMSEP) and the coefficient of determination (R²). A model is considered valid if the RMSEP is sufficiently low and the R² is high (e.g., >0.98) for the intended purpose.
- Routine Prediction: For future unknown tablets, a single NIR spectrum is collected and the validated PLS model is applied to instantly predict the API content.
Diagram 1: NIR Quantitative Analysis Workflow
The Scientist's Toolkit: Essential Reagents and Materials
Successful implementation of NIR spectroscopy requires both hardware and software components. The following table details the key solutions and materials essential for conducting NIR analysis in a pharmaceutical context.
Table 2: Essential Research Reagent Solutions for NIR Pharmaceutical Analysis
| Item / Solution | Function / Purpose | Technical Specification Notes |
|---|---|---|
| NIR Spectrometer | Measures absorption/reflection of NIR light by the sample. | Types: FT-NIR (high precision), Benchtop (workhorse), Portable/Handheld (on-site). MEMS-based systems are emerging [46]. |
| Fiber Optic Probe | Enables remote, in-line, or on-line measurements in process environments. | Allows separation of spectrometer from sample; essential for PAT [42] [45]. |
| Chemometrics Software | Extracts meaningful information from complex, multivariate NIR spectra. | Must include algorithms for PCA (qualitative) and PLS (quantitative) analysis [42] [43]. |
| Calibration Standards | For instrument performance verification and quantitative model development. | Certified reference materials with known properties (e.g., API concentration, polymorphic form) [42]. |
| Spectral Library | Database of reference spectra for rapid identification of raw materials. | Built using multiple batches of authenticated materials; used with PCA or similar models [42]. |
Regulatory Considerations and Future Outlook
The use of NIR spectroscopy in the highly regulated pharmaceutical world requires careful attention to validation and regulatory compliance. Regulatory bodies like the U.S. Food and Drug Administration (FDA) have provided frameworks, such as the PAT guidance, which encourage the use of advanced analytical technologies like NIR for real-time quality assurance [42] [47] [46]. Method validation is critical and must demonstrate that the NIR method is fit-for-purpose in terms of specificity, accuracy, precision, and robustness, following the principles outlined in compendial chapters such as the USP general chapter on near-infrared spectrophotometry [42]. The future of NIR spectroscopy in pharmaceuticals is closely tied to the industry's move toward continuous manufacturing [47] [46]. The ability of NIR to provide real-time, non-destructive data makes it the ideal PAT tool for monitoring and controlling these continuous processes, enabling real-time release and enhancing overall manufacturing agility. Furthermore, advancements in NIR imaging techniques, which combine NIR spectroscopy with digital image processing, provide spatial distribution of components within a solid dosage form, opening new avenues for understanding product homogeneity and stability [42]. The ongoing development of more robust, portable, and cost-effective instrumentation will further democratize this powerful technology, solidifying its role as an indispensable tool in modern pharmaceutical analysis.
Diagram 2: NIR Spectroscopy Principle and Data Flow
Radiopharmaceuticals represent a cutting-edge class of therapeutic agents that enable the precise delivery of radionuclides to targeted disease lesions through specialized targeting vectors. These sophisticated compounds have revolutionized nuclear medicine by allowing simultaneous diagnosis and treatment of various diseases, particularly in oncology. The fundamental principle underlying radiopharmaceutical function involves the local deposition of radionuclides within targeted cells, causing systematic and irreparable damage through DNA disruption while minimizing impact on healthy tissues [48]. This targeted approach stands in stark contrast to conventional radiotherapy by restricting radiation effects primarily to pathological cells, thereby significantly reducing normal organ injury [48].
The integration of radiopharmaceuticals with advanced imaging modalities including positron emission tomography (PET) and single-photon emission computed tomography (SPECT) has enabled comprehensive monitoring of whole-body disease lesions, facilitating accurate patient stratification noninvasively [48]. The remarkable advantage of radiopharmaceuticals lies in their ability to visualize and quantify drug accumulation within lesions, allowing clinicians to implement personalized treatment strategies based on real-time assessment of therapeutic agents [48]. This paradigm, known as radiotheranostics, combines precise diagnostic capabilities with efficient targeted radiopharmaceutical therapy (RPT), representing a significant advancement in precision medicine.
The physical foundation of radiopharmaceutical operation connects directly to electron energy level transitions and atomic spectroscopy. When radionuclides decay, they emit radiation through processes that involve electron transitions between atomic energy levels. In diagnostic applications, characteristic X-rays are produced when outer-shell electrons fill vacancies in inner shells, emitting electromagnetic radiation with energies corresponding to specific electron transitions [30]. Similarly, in therapeutic applications, the energy deposited in tissues results from various decay processes that ultimately cause ionization and DNA damage through electron interactions. The spectroscopic properties of these transitions provide the fundamental basis for both imaging and therapeutic effects in nuclear medicine.
Electron Transitions and Radiopharmaceutical Mechanisms
Atomic Spectroscopic Foundations
The operation of radiopharmaceuticals is fundamentally rooted in the principles of atomic spectroscopy and electron energy transitions. When radionuclides decay, they initiate cascades of electron transitions that ultimately produce detectable signals or therapeutic effects. In the X-ray fluorescence process, the excitation energy must equal or exceed the binding energy required to remove an electron from its inner shell (E ≥ EB) [30]. Subsequent electron transitions from outer to inner shells produce characteristic X-rays that enable imaging and detection. This process forms the basis for numerous diagnostic applications in nuclear medicine.
The energy scales involved in these transitions vary significantly depending on the electron shells involved. For instance, in magnesium atoms, the binding energy of K-shell electrons is approximately three orders of magnitude greater than optical emission spectroscopy ionization-excitation energies [30]. This substantial energy differential explains why X-ray emissions provide sufficient penetration for medical imaging while optical emissions do not. The precise energy signatures emitted during electron transitions serve as fingerprints for specific elements and compounds, allowing accurate tracking of radiopharmaceutical distribution within the body.
Therapeutic Mechanisms at the Cellular Level
Radiopharmaceutical therapy induces cell death primarily through radiation-induced DNA damage. The cytotoxic effect and its extent on surrounding cells depend critically on the physical properties of the radionuclide employed [49]. Two primary classes of therapeutic radionuclides exist: those emitting penetrating radiation (photons) used primarily for imaging, and those emitting non-penetrating radiation (electrons and alpha particles) used for therapy [49]. The latter category includes beta emitters, alpha emitters, and Auger electron emitters, each with distinct biological effectiveness profiles.
Alpha particles and Auger electrons, characterized by higher linear energy transfers (LET), deposit their energy over shorter distances, resulting in increased cell death and limited repair of DNA damage [49]. The efficiency of specific radionuclides depends on their energy characteristics and penetration depth into tissues. Optimal therapeutic outcomes require matching the emission range of radioactive particles to target size, ensuring maximal dose delivery to pathological tissues while minimizing damage to adjacent healthy structures [49]. This precise targeting represents the cornerstone of modern radiopharmaceutical design and application.
Radionuclides in Nuclear Medicine
Classification and Properties
Radionuclides employed in nuclear medicine demonstrate diverse physical and biochemical characteristics including variable half-lives, decay modes, radiation energies, and retention properties in target tissues [48]. The International Commission on Radiological Protection (ICRP) identifies approximately 1,200 radionuclides, yet only several dozen have established roles in clinical and scientific applications [48]. These radionuclides are systematically categorized based on their decay properties and clinical applications, with optimal selection depending on specific diagnostic or therapeutic requirements.
Table 1: Classification of Radionuclides in Nuclear Medicine
| Category | Decay Mode | Primary Applications | Representative Radionuclides | Key Characteristics |
|---|---|---|---|---|
| Single-Photon Emitters | Gamma emission | SPECT imaging | Technetium-99m, Iodine-123 | Gamma energy ~140-159 keV; Half-life: 6-13 hours |
| Positron Emitters | Positron emission | PET imaging | Fluorine-18, Gallium-68, Carbon-11 | Positron range 0.6-2.6 mm; Half-life: 20 minutes to 68 hours |
| Beta Emitters | Beta decay | Therapeutic applications | Lutetium-177, Iodine-131, Yttrium-90 | Medium penetration (0.2-12 mm); Half-life: 2.7-8 days |
| Alpha Emitters | Alpha decay | Therapeutic applications | Actinium-225, Astatine-211, Lead-212 | High LET (50-230 keV/μm); Short range (40-100 μm) |
| Auger Electron Emitters | Electron capture | Therapeutic applications | Iodine-125, Indium-111 | Very short range (<10 μm); High LET near decay site |
Clinically Significant Radionuclides
Technetium-99m deserves particular emphasis among single-photon emitters due to its predominant role in approximately 80% of SPECT procedures [48]. This widespread utilization stems from its favorable gamma-ray energy of 140.5 keV, which balances detection efficiency with minimal toxicity risk, coupled with an optimal 6-hour half-life that allows sufficient time for procedures while limiting patient radiation exposure [48]. Among positron emitters, Fluorine-18 has emerged as particularly valuable for PET applications due to its low positron range that enables high spatial resolution imaging and its clear positron emission profile (97% positron emission, 3% electron capture) [48].
Therapeutic radionuclide selection requires careful consideration of multiple factors. Appropriate physical half-life represents a crucial parameter, with optimal ranges typically falling between 6 hours and 10 days to balance effective treatment duration with radiation safety concerns [48]. Additionally, therapeutic radionuclides must emit high linear energy transfer (LET) radiation to efficiently kill target cells [48]. Lutetium-177 has gained significant clinical traction due to its ideal properties for therapy, including medium-energy beta emissions, manageable half-life of 6.7 days, and the presence of gamma photons that permit simultaneous imaging during treatment [49].
Radiopharmaceutical Design and Components
Structural Elements
Modern radiopharmaceuticals comprise three essential components: a radioisotope with associated chelator, a targeting agent, and a linker that connects these elements [49]. This modular design enables precise customization for specific clinical applications. The targeting component, which may include small molecules, peptides, or antibodies, determines the biodistribution and specificity of the radiopharmaceutical by recognizing specific molecular markers on target cells [48]. The radionuclide component provides the diagnostic or therapeutic effect, while the linker influences pharmacokinetic properties and in vivo stability.
Recent advancements have focused on optimizing each component to enhance clinical performance. Targeting vectors with improved binding affinity to disease-specific targets have been developed through sophisticated modification strategies [48]. Simultaneously, novel chelation chemistry has emerged to enhance radiolabeling efficiency and in vivo stability [48]. These refinements have yielded radiopharmaceuticals demonstrating superior tumor uptake, prolonged retention time, and favorable pharmacokinetic profiles aligned with clinical requirements [48].
Theranostic Pairs
The radiotheranostic paradigm employs matched radionuclide pairs for simultaneous diagnosis and treatment. These pairs share identical targeting vectors but incorporate different radionuclides optimized for their respective roles. Prominent examples include Gallium-68 and Lutetium-177 paired with somatostatin analogs ([⁶⁸Ga]Ga-DOTA-TATE and [¹⁷⁷Lu]Lu-DOTA-TATE) for neuroendocrine tumors, and similarly paired prostate-specific membrane antigen (PSMA) ligands ([⁶⁸Ga]Ga-PSMA-11 and [¹⁷⁷Lu]Lu-PSMA-617) for prostate cancer [48]. This approach enables precise patient selection, treatment planning, and therapy response assessment using biologically identical compounds.
Table 2: FDA-Approved Radiotheranostic Pairs
| Theranostic System | Diagnostic Agent | Therapeutic Agent | Primary Indication | Key Clinical Trial Findings |
|---|---|---|---|---|
| Somatostatin Receptor | [⁶⁸Ga]Ga-DOTA-TATE (Netspot) | [¹⁷⁷Lu]Lu-DOTA-TATE (Lutathera) | Neuroendocrine tumors | Progression-free survival: 65.2% (treatment) vs 10.8% (control) at 20 months |
| PSMA-Targeting | [⁶⁸Ga]Ga-PSMA-11 (Locametz) | [¹⁷⁷Lu]Lu-PSMA-617 (Pluvicto) | Metastatic castration-resistant prostate cancer | PSA reduction ≥50%: 46% (treatment) vs 7% (control); Overall survival: 15.3 vs 11.3 months |
| Radium-223 Dichloride | N/A | Radium-223 (Xofigo) | Bone metastases from prostate cancer | Overall survival: 14.9 vs 11.3 months; Time to skeletal event: 15.6 vs 9.8 months |
Experimental Protocols and Methodologies
Radiopharmaceutical Development Workflow
The development of novel radiopharmaceuticals follows a systematic pathway from target identification to clinical implementation. Initial target validation confirms the presence and specificity of molecular markers on pathological cells. Subsequent ligand design focuses on creating targeting vectors with high affinity and specificity for the identified markers. Radiolabeling optimization establishes efficient methods for incorporating radionuclides while maintaining biological activity. Preclinical evaluation assesses binding affinity, internalization efficiency, and cytotoxicity in relevant cellular and animal models. Finally, clinical translation involves dosimetry studies, toxicity assessment, and efficacy evaluation in human subjects.
Dosimetry Protocol for ¹⁷⁷Lu-Based Therapies
Accurate dosimetry represents a critical component of radiopharmaceutical therapy to balance efficacy and safety. The following protocol outlines standardized methodology for patient-specific dosimetry in ¹⁷⁷Lu-based treatments:
Imaging Protocol:
- Acquire quantitative SPECT/CT images at multiple time points post-injection (typically 4, 24, 72, and 144 hours)
- Maintain consistent imaging geometry and reconstruction parameters across all time points
- Perform camera calibration using standardized sources to ensure quantitative accuracy
- Obtain blood and urine samples for bioassay validation at each imaging time point
Image Processing:
- Reconstruct SPECT images using iterative reconstruction with attenuation, scatter, and collimator response corrections
- Segment organs of interest (tumors, kidneys, liver, spleen, bone marrow) on CT images
- Register sequential SPECT images to account for patient movement between scans
- Calculate time-integrated activity coefficients using exponential curve fitting to time-activity data
Dose Calculation:
- Apply appropriate organ-level or voxel-level dose calculation methods
- Utilize Monte Carlo simulations or dose point kernel convolution techniques
- Calculate absorbed doses for tumors and organs at risk (mean dose for organs, minimum dose for tumors)
- Compare results to established tolerance doses (kidneys: 23 Gy, bone marrow: 1.5 Gy, liver: 25 Gy)
Activity Administration Adjustment:
- Based on calculated absorbed doses, adjust subsequent treatment activities to maximize tumor dose while maintaining organ doses below tolerance limits
- Implement fractionated approach for treatments requiring multiple cycles
- Document all dosimetry results for treatment planning and regulatory compliance
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential Research Reagents for Radiopharmaceutical Development
| Category | Specific Reagents | Function | Application Notes |
|---|---|---|---|
| Targeting Vectors | Peptides (DOTATATE, PSMA-617), monoclonal antibodies, small molecules | Provides target specificity and binding affinity | Determine pharmacokinetics and tumor uptake; Critical for therapeutic index |
| Radionuclides | ¹⁷⁷Lu, ⁶⁸Ga, ⁹⁰Y, ²²⁵Ac, ⁹⁹ᵐTc, ¹¹¹In, ¹⁸F | Source of radiation for imaging or therapy | Selection based on decay properties, half-life, and emission characteristics |
| Chelators | DOTA, NOTA, DTPA, HBED | Coordinates radionuclides to targeting vectors | Impacts stability, pharmacokinetics, and labeling efficiency |
| Quality Control Materials | Radio-TLC, radio-HPLC, instant thin-layer chromatography | Verifies radiochemical purity and stability | Essential for regulatory compliance and batch consistency |
| Cell Culture Models | Target-positive and target-negative cell lines | In vitro assessment of binding and internalization | Validates target specificity and determines binding affinity |
| Animal Models | Xenograft models, genetically engineered models, patient-derived xenografts | Preclinical efficacy and biodistribution studies | Predicts human performance and optimizes dosing regimens |
| Imaging Phantoms | Sphere phantoms, organ-specific phantoms | Quantification and calibration of imaging systems | Ensures accurate activity quantification for dosimetry |
Dosimetry and Treatment Planning
Principles of Radiopharmaceutical Dosimetry
Dosimetry in radiopharmaceutical therapy focuses on quantifying energy deposition in target tissues and organs at risk. The absorbed dose, measured in Gray (joules per kilogram), serves as the primary determinant of biological effects including radiation-induced cell death [49]. Accurate dosimetry requires comprehensive understanding of radiopharmaceutical pharmacokinetics, including cellular uptake, retention time, non-uniform distribution within targets, and metabolic behavior [49]. These patient-specific biokinetic data fundamentally influence dose calculations and subsequent treatment optimization.
The paradigm for RPT administration is shifting from fixed empirical dosing to patient-specific dosimetry-based activity determination [49]. This transition acknowledges the significant interpatient variability observed in radiopharmaceutical biodistribution and retention. Factors including tumor burden, receptor density, renal function, and hepatic metabolism substantially influence radiation dose delivery, necessitating individualized treatment approaches to maximize therapeutic efficacy while minimizing toxicity [49]. The European Council Directive 2013/59/Euratom now mandates justification of medical exposure to ionizing radiation and emphasizes optimization through individualized treatment planning [49].
Comparative Dosimetry: RPT versus EBRT
Fundamental differences exist between radiopharmaceutical therapy and external beam radiotherapy in dosimetry approach and implementation. RPT employs continuous, gradually decreasing radiation dose rates, resulting in complex temporal and spatial dose distributions compared to EBRT's fractionated high dose rate approach [49]. Additionally, EBRT administers consistent absorbed dose per cell regardless of cell number, while in RPT, the absorbed dose per cell from internal emissions depends on emission ranges and the quantity of targeted cells [49]. These distinctions necessitate specialized methodologies for RPT dosimetry that account for evolving activity distributions over time.
Emerging Applications and Future Directions
Expanding Clinical Applications
While radiotheranostics have established robust applications in oncology, their implementation is expanding to diverse medical fields including neurodegenerative diseases, cardiovascular diseases, and inflammatory conditions [48]. In neurology, radiolabeled compounds targeting amyloid-beta and tau proteins enable early diagnosis and treatment monitoring for Alzheimer's disease. Cardiology applications include radiolabeled agents for identifying vulnerable atherosclerotic plaques and assessing myocardial viability. Inflammation imaging utilizes radiopharmaceuticals that target specific immune cells or molecular processes involved in inflammatory pathways.
The integration of radiopharmaceutical therapy with other treatment modalities represents another promising direction. Combination approaches linking RPT with immunotherapy, chemotherapy, or external beam radiotherapy demonstrate potential synergistic effects [49]. For instance, radiation can enhance tumor immunogenicity, potentially improving response to immunotherapeutic agents. Similarly, RPT combined with EBRT may improve local control while addressing disseminated disease, particularly in challenging malignancies like prostate cancer and hepatocellular carcinoma [49].
Novel Radionuclides and Targeting Strategies
Therapeutic radionuclides with higher linear energy transfer and longer half-lives are emerging as the field shifts emphasis from diagnostic imaging to targeted therapy [48]. Alpha-emitters including actinium-225, astatine-211, and lead-212 demonstrate particular promise due to their high anti-tumor efficacy and minimal normal tissue toxicity [48]. These radionuclides deposit substantial energy over extremely short distances (40-100 μm), causing complex DNA damage that is difficult for cells to repair. Clinical trials evaluating alpha-emitting radiopharmaceuticals have demonstrated robust anti-tumor effects across various cancer types [48].
Advanced targeting strategies are revolutionizing radiopharmaceutical design. Novel ligand formats including minibodies, nanobodies, and affibodies offer improved pharmacokinetic profiles compared to conventional antibodies. Pretargeting approaches that separate targeting vector administration from radionuclide delivery enhance tumor-to-background ratios. Additionally, theranostic nanoparticles incorporating both imaging and therapeutic components enable multimodal treatment approaches with real-time monitoring capabilities. These innovations collectively expand the potential of radiopharmaceuticals to address increasingly challenging clinical scenarios.
Radiopharmaceuticals and theranostics represent a transformative approach in modern medicine, leveraging fundamental principles of electron transitions and atomic spectroscopy to achieve precise disease targeting. The integration of diagnostic and therapeutic capabilities within unified platforms enables unprecedented personalization of treatment based on individual patient characteristics and real-time assessment of therapeutic response. Continued advancements in radionuclide production, targeting vector design, and dosimetry methodology will further enhance the precision and efficacy of these powerful agents. As research expands into novel applications beyond oncology and develops increasingly sophisticated theranostic pairs, radiopharmaceuticals are poised to play an increasingly vital role in precision medicine across diverse therapeutic areas.
Organic photodetectors (OPDs) are emerging as leading candidates for next-generation image sensors due to their tunable photophysical properties, which enable broadband detection from the visible to X-ray regimes. These devices open new possibilities for intelligent systems such as fingerprint sensing, gesture recognition, and medical imaging [50]. Unlike traditional inorganic semiconductors, OSCs possess unique advantages including mechanical flexibility, cost-effective processing, and the ability to tailor their optical bandgap through chemical modifications of their molecular structure. This enables detection across ultraviolet (UV), visible (VIS), and near-infrared (NIR) spectral regions without requiring external filters [51].
The fundamental operation of OPDs relies on electron energy level transitions within π-conjugated organic molecules. In organic semiconductors (OSCs), the valence band is represented by the highest occupied molecular orbital (HOMO) and the conduction band by the lowest unoccupied molecular orbital (LUMO). Unlike inorganic semiconductors that generate free charge carriers upon photoexcitation, OSCs produce bound electron-hole pairs known as excitons [51]. The energy gap between HOMO and LUMO levels typically ranges from 1.5 to 3.0 eV, corresponding to the spectral range from visible to near-infrared light [51].
Fundamental Operating Principles
Photodetection Mechanism in Organic Semiconductors
The photodetection process in OPDs occurs through several sequential steps:
Photon Absorption and Exciton Generation: When light with energy exceeding the HOMO-LUMO gap illuminates the organic active layer, electrons are promoted from the ground state to a higher energy excited state [22]. This process creates bound electron-hole pairs (excitons) with typical binding energies of 0.1-0.5 eV [51].
Exciton Diffusion: The photogenerated excitons diffuse through the organic material toward a donor-acceptor (D/A) interface. The diffusion length in disordered OSCs is typically 5-20 nm, which constrains the active layer thickness in efficient devices [51].
Charge Separation: At the D/A interface, the excitons dissociate into free charge carriers (electrons and holes). This process is driven by energy level offsets between donor and acceptor materials, typically requiring 0.3-0.5 eV for efficient charge separation [52].
Charge Transport and Collection: The separated electrons and holes transport through their respective percolation pathways to the electrodes, where they are collected as photocurrent [51].
Three distinct charge separation channels exist in OPDs: electron transfer, hole transfer, and energy transfer-induced electron/hole transfer. Recent studies on non-fullerene acceptor-based blends have confirmed that charge separation can occur under nearly zero driving force in optimized OPV systems, with specific molecular packing patterns playing an important role in facilitating this process [52].
Key Performance Parameters
The performance of OPDs is characterized by several critical parameters:
- Responsivity (R): The ratio of photocurrent generated per unit power of incident light.
- Specific Detectivity (D*): The ability to detect weak optical signals, considering noise current.
- Dark Current Density (Jd): The current flowing through the device in the absence of light, which sets the noise floor.
- Linear Dynamic Range (LDR): The range over which the photocurrent response remains linear with light intensity.
- External Quantum Efficiency (EQE): The ratio of collected charge carriers to incident photons.
Table 1: Key Performance Parameters of State-of-the-Art OPDs
| Parameter | Typical Range | Advanced Performance | Measurement Conditions |
|---|---|---|---|
| Dark Current Density (Jd) | 10-6 to 10-9 A/cm² | < 10-10 A/cm² | Reverse bias: 1-5 V [50] |
| Specific Detectivity (D*) | 1011 to 1013 Jones | > 1013 Jones | 500-800 nm, 0-2 V bias [50] |
| Linear Dynamic Range (LDR) | 60-100 dB | 120-150 dB | White light illumination [51] |
| Response Speed | 100 ns - 10 μs | < 100 ns | Pulsed laser measurement [51] |
| External Quantum Efficiency | 50-80% | > 90% | At peak wavelength [51] |
Noise Suppression and Dark Current Management
Origins and Mechanisms of Noise in OPDs
Minimizing dark current density (Jd) is paramount for OPD performance as it establishes the noise floor. Thermal and low-frequency noises further depend on the shunt resistance and trap dynamics, thereby constraining the signal-to-noise ratio, dynamic range, and specific detectivity (D*) [50]. The microscopic origins of Jd can be distinguished through two universal leakage channels:
- Bulk Thermal Generation: Charge carriers generated via mid-gap or tail-state traps contribute to shunt paths within the active layer [50].
- Field-Assisted Carrier Injection: Carriers injected through interfacial barriers at electrode-organic interfaces under applied bias [50].
Beyond white-noise considerations, low-frequency (1/f) noise critically impacts stability and detectivity. Its origins in organic semiconductors remain debated but are associated with charge trapping/detrapping processes and molecular reorientations [50].
Strategies for Noise Suppression
Recent progress in noise suppression has focused on three primary approaches:
- Active-Layer Design: Engineering donor-acceptor systems with optimized energy level alignment to reduce charge recombination while maintaining efficient charge separation [50].
- Disorder Management: Controlling the density of trap states through material purification and optimized processing conditions to minimize tail-state formation [50] [51].
- Interlayer Engineering: Introducing specialized buffer layers between electrodes and the active layer to block unwanted charge injection [50].
Advanced doping strategies have proven effective in managing dark current. For instance, rubrene doping into the p-layer of p-i-n OPDs has demonstrated significant improvements in open-circuit voltage (VOC) and fill factor (FF), similar to doping effects observed in organic light-emitting diodes [53]. The energy level alignment at organic semiconductor-electrode interfaces critically determines charge injection barriers. The density of states in the organic semiconductor serves as a key factor, with its shape and the energy distribution of electronic states tailing into the fundamental gap determining both the minimum value of practically achievable injection barriers and their spatial profile [54].
Material Systems and Device Architectures
Organic Semiconductor Materials
OSCs are categorized as small molecules, oligomers (few monomer units), and polymers (many monomer units). They are characterized by relatively low energy (≈10 kcal/mol) van der Waals intermolecular bonds compared to Si-Si (78 kcal/mol) covalent bonds. This mild propensity to form ordered structures enables deposition using low-cost solution-based techniques [51].
Table 2: Representative Organic Semiconductor Materials for OPDs
| Material Class | Example Compounds | HOMO-LUMO Gap (eV) | Spectral Response | Key Advantages |
|---|---|---|---|---|
| Small Molecules | Rubrene, ZnPc, C₆₀ | 1.8 - 2.5 | Visible to NIR | High purity, well-defined structure [53] |
| Polymer Donors | P3HT, PBDB-T, PBDTTT-C-T | 1.7 - 2.1 | Visible | Solution processability, mechanical flexibility [55] |
| Non-Fullerene Acceptors | ITIC, Y6, L-series | 1.3 - 1.8 | Visible to NIR | Strong absorption, tunable energy levels [52] |
| Transparent Electrodes | PEDOT:PSS, doped graphene | N/A | N/A | High transparency, solution processability [55] |
Device Architectures and Processing Techniques
The most prevalent deposition techniques for OSC thin films include vacuum evaporation and spin coating. Vacuum evaporation preserves OSC purity by preventing modification from water and oxygen, while spin coating offers straightforward processing for creating thick OSC thin films [51].
Conventional Architecture: Glass/ITO/PEDOT:PSS/Active Layer/Metal Cathode
- ITO provides high transparency and conductivity
- PEDOT:PSS serves as hole transport layer
- Active layer typically consists of bulk heterojunction blend
- Metal cathode (Al, Ag) for charge collection
Inverted Architecture: Glass/ITO/Electron Transport Layer/Active Layer/Hole Transport Layer/Transparent Electrode
- Improved device stability
- Compatibility with high-work-function transparent electrodes
- Reduced degradation at the organic-electrode interface
Advanced Architectures: Recent developments include semitransparent OPDs using solution-doped graphene electrodes, which provide energetically favorable band alignment for carrier extraction while maintaining high transparency across visible wavelengths. These devices can achieve power conversion efficiency of 4.2%, representing 85.7% of the PCE of control devices based on metallic reflecting electrodes [55].
Experimental Characterization Methods
Electronic Spectroscopy for OPD Characterization
Electronic spectroscopy techniques are essential for characterizing OPD materials and devices. These methods rely on the quantized nature of energy states, where electrons excited from their initial ground state briefly exist in a higher energy excited state before relaxing back to their original stable state and releasing energy as photons [22].
Current Density-Voltage (J-V) Measurements:
- Procedure: Sweep voltage from reverse to forward bias while measuring current under dark and illuminated conditions
- Data extracted: Dark current density, open-circuit voltage (VOC), short-circuit current (JSC), fill factor (FF)
- Equipment: Source measure unit, calibrated light source, environmental-controlled probe station
External Quantum Efficiency (EQE) Spectroscopy:
- Procedure: Illuminate device with monochromatic light while measuring short-circuit current
- Data extracted: Spectral response, charge collection efficiency
- Equipment: Monochromator, lock-in amplifier, reference photodetector
Impedance Spectroscopy:
- Procedure: Apply small AC voltage bias across frequency range (typically 1 Hz - 1 MHz)
- Data extracted: Shunt and series resistance, chemical capacitance, recombination lifetime
- Equipment: Impedance analyzer, environmental-controlled sample holder
Ultraviolet Photoelectron Spectroscopy (UPS):
- Procedure: Irradiate sample with UV photons (He I: 21.2 eV or He II: 40.8 eV) and analyze kinetic energy of emitted electrons
- Data extracted: Work function, HOMO level position, ionization energy
- Equipment: Ultra-high vacuum system, UV source, electron energy analyzer [54] [55]
Research Reagent Solutions and Materials
Table 3: Essential Research Materials for OPD Development
| Material/Reagent | Function | Example Specifications | Application Notes |
|---|---|---|---|
| PEDOT:PSS | Hole transport layer, electrode doping | Clevios PH1000, sheet resistance: 70-100 Ω/sq | Spin-coating at 3000-5000 rpm, anneal at 120-150°C [55] |
| P3HT | Polymer donor | RR > 98%, Mw: 30-60 kDa | Solution concentration: 10-20 mg/mL in chlorobenzene [55] |
| PCBM | Fullerene acceptor | >99.5% purity | Donor:acceptor ratio typically 1:1 to 1:1.5 [55] |
| Non-Fullerene Acceptors (e.g., L4, L5) | High-performance acceptor | Custom synthesis | Enhanced NIR response, reduced voltage loss [52] |
| MoOₓ | Hole transport layer | Thermal evaporation: 5-10 nm | Work function ~5.3 eV, improves hole injection [55] |
| Doped Graphene | Transparent electrode | 1-5 layers, sheet resistance: 50-300 Ω/sq | Polymer-free transfer, PEDOT doping for WF tuning [55] |
| Rubrene | Dopant for mobility enhancement | Sublimed grade, >99% purity | Vacuum evaporation, concentration: 1-10 wt% [53] |
Advanced Applications and Future Perspectives
OPDs have found applications across multiple fields including medical imaging, gesture recognition, fingerprint sensing, and environmental monitoring. Their unique properties enable novel implementations not feasible with conventional inorganic photodetectors [50]. The mechanical conformability of organic materials inherently provides advantages for developing large-area, flexible, wearable photodetectors suited for diverse and evolving future needs [51].
Future research directions focus on several key areas:
- Reducing Dark Current: Roadmaps toward sub-femtoampere Jd through advanced material design and interface engineering [50].
- Enhancing Detectivity: Optimizing noise suppression strategies to achieve D* values exceeding 1013 Jones across visible and NIR spectra [50].
- Improving Stability: Addressing OSC vulnerability to environmental degradation through advanced encapsulation and stable material design [51].
- Narrowband Detection: Developing OPDs with intrinsic spectral selectivity for color-specific applications without external filters [51].
The continued advancement of OPD technology relies on fundamental research into electron energy level transitions and their relationship to material structure and device performance. As understanding of these relationships deepens, organic photodetectors are poised to expand their applications in sensing and imaging technologies, potentially surpassing the capabilities of conventional inorganic devices in specific application domains.
Core-Level Spectroscopy (XAS, RIXS) for Probing Electronic Structure
Core-level spectroscopy encompasses a suite of advanced analytical techniques that utilize X-rays to probe the electronic structure and local atomic environment of materials. These techniques are foundational to modern materials science, chemistry, and pharmacology, providing element-specific insights that are indispensable for understanding functional properties at the atomic and molecular levels. X-ray Absorption Spectroscopy (XAS) and Resonant Inelastic X-ray Scattering (RIXS) represent two powerful methods in this arsenal, enabling researchers to investigate electronic configurations, oxidation states, coordination geometries, and chemical bonding. The unique capability of these techniques to analyze solids, liquids, and gases without extensive sample preparation makes them particularly valuable for studying complex systems, including pharmaceutical compounds, energy storage materials, and quantum materials [33] [56].
The fundamental physical process underlying these techniques involves the interaction of X-ray photons with core-level electrons of specific elements. When incident X-ray energy matches the binding energy of a core electron (e.g., in the 1s, 2s, or 2p orbital), the electron is excited to unoccupied states or ejected into the continuum, creating a core hole. The subsequent relaxation processes, which may involve radiative (fluorescence) or non-radiative (Auger) transitions, produce measurable signals that contain detailed information about the electronic structure and local environment of the absorbing atom [33]. This technical guide explores the principles, methodologies, and applications of XAS and RIXS, framed within the broader context of electron energy level transitions spectroscopy research, to provide researchers and drug development professionals with a comprehensive resource for electronic structure characterization.
Theoretical Foundations
Fundamental Physical Principles
Core-level spectroscopy techniques are based on the photoelectric effect, where an incident X-ray photon is absorbed by an atom, ejecting a core electron. The probability of this absorption event is quantified by the absorption coefficient (μ), which exhibits sharp increases known as absorption edges when the photon energy reaches the binding energy of specific core electrons. These edges are labeled according to the principal quantum number of the shell from which the electron is ejected: K-edge (1s electron), L-edge (2s or 2p electrons), and M-edge (3s, 3p, or 3d electrons) [33] [56].
The excitation process creates an unstable state with a core hole, which rapidly decays through fluorescence or Auger processes. In fluorescence decay, an electron from a higher energy level fills the core hole, emitting an X-ray photon with energy characteristic of the specific transition. The energies and probabilities of these processes are exquisitely sensitive to the chemical environment of the atom, including oxidation state, coordination symmetry, and bond character, making them powerful probes of electronic structure [33] [57].
Relationship Between Techniques
XAS and RIXS are complementary techniques that probe related but distinct aspects of electronic structure. XAS primarily measures the transition of core electrons to unoccupied states, providing information about the density of empty states, oxidation state, and local symmetry. RIXS, in contrast, involves resonant excitation followed by measurement of the emitted photon, offering insights into electronic excitations, charge transfer, and multiplet structures with enhanced energy resolution compared to conventional XAS [58] [57].
Table 1: Comparison of Core-Level Spectroscopy Techniques
| Technique | Primary Process | Information Obtained | Element Selectivity | Key Advantages |
|---|---|---|---|---|
| XAS | Core electron excitation to unoccupied states | Oxidation state, local symmetry, unoccupied density of states | Yes | Direct quantitative analysis, suitable for concentrated samples |
| XES | Decay of core hole with photon emission | Occupied density of states, chemical bonding | Yes | Complementary to XAS, probes occupied states |
| RIXS | Resonant excitation followed by emission | Low-energy excitations, charge transfer, multiplet structures | Yes | Enhanced resolution, Raman-like capabilities, bulk sensitive |
The relationship between these techniques can be understood through a unified theoretical framework where XAS represents the initial excitation process, while XES and RIXS probe the decay channels. When performed with tunable incident energy, XES becomes resonant (RIXS), enabling the study of electronic excitations at energy transfers much lower than the incident X-ray energy, in some cases as low as a few meV for probing phonon excitations [57].
X-Ray Absorption Spectroscopy (XAS)
X-ray Absorption Spectroscopy is a versatile technique for probing the local electronic and structural environment of specific elements in matter. The XAS spectrum is typically divided into three main regions: the absorption threshold, which reveals the lowest unoccupied states; the X-ray Absorption Near-Edge Structure (XANES), which extends approximately 10-150 eV above the threshold and contains information about electronic structure, oxidation state, and coordination geometry; and the Extended X-ray Absorption Fine Structure (EXAFS), which appears at higher energies and provides quantitative information about bond distances, coordination numbers, and structural disorder [33] [56].
A key advantage of XAS is its element specificity, achieved by tuning the incident X-ray energy to the characteristic absorption edge of the element of interest. This allows researchers to probe specific elements even in complex multicomponent systems, without interference from the matrix. Additionally, XAS does not require long-range order, making it applicable to crystalline, amorphous, and liquid samples alike [33].
Experimental Methodologies
XAS measurements are predominantly performed at synchrotron radiation facilities, which provide the intense, tunable X-ray beams necessary for these experiments. Three primary detection modes are employed, each with distinct advantages for different sample types:
Transmission Mode: Measures the intensity of X-rays before (I0) and after (It) passing through the sample using ionization chambers. This method is ideal for homogeneous samples with relatively high concentrations (>10%) of the element of interest and provides high-quality spectra with short acquisition times [33].
Fluorescence Mode: Detects the characteristic X-rays emitted during the decay of the core hole. The incident beam intensity (I0) is recorded with an ionization chamber, while the fluorescence signal (If) is measured with a dedicated detector, typically arranged at 90° geometry to minimize elastic scattering. This mode is essential for dilute systems or samples with low concentrations of the target element [33].
Electron Yield Mode: Measures the electrons emitted as a result of the absorption process. This method is particularly surface-sensitive and useful for thin films or surface studies [33].
The following workflow diagram illustrates the key decision points in planning and executing a XAS experiment:
Data Interpretation and Analysis
XANES analysis focuses primarily on the absorption edge position, which shifts to higher energies with increasing oxidation state due to the increased binding energy of core electrons. For example, the Mn K-edge shifts by approximately 1-2 eV per oxidation state change in manganese oxides [59]. The pre-edge features provide information about coordination geometry and symmetry-forbidden transitions, while the edge shape and white line intensity reflect the density of unoccupied states and covalent mixing with ligand orbitals [33] [56].
EXAFS analysis involves Fourier transformation of the oscillatory signal extending from approximately 30-1000 eV above the absorption edge. The resulting radial distribution function provides interatomic distances, coordination numbers, and disorder parameters (Debye-Waller factors). Quantitative EXAFS analysis requires fitting theoretical models to experimental data, using programs such as ATHENA, to extract precise structural parameters [33] [56].
Resonant Inelastic X-Ray Scattering (RIXS)
Resonant Inelastic X-ray Scattering is an advanced photon-in/photon-out technique that combines element specificity with enhanced energy resolution for studying electronic excitations. In RIXS, the incident X-ray energy is tuned to a specific absorption resonance, creating a core-excited intermediate state. The subsequent decay of this state produces emitted photons whose energy distribution is analyzed to reveal detailed information about low-energy excitations, including d-d excitations, charge transfers, magnons, and phonons [58] [57].
A distinctive feature of RIXS is its ability to measure excitations with energy transfers much lower than the incident X-ray energy, in some cases as low as a few meV. This makes it possible to probe the electronic structure of materials with bulk sensitivity and without the vacuum requirements of soft X-ray techniques. The resonance enhancement provides elemental selectivity, while the energy transfer analysis enables the separation of different excitation channels [57].
Experimental Considerations
RIXS experiments require synchrotron radiation sources with high brightness and energy resolution. The instrumental energy bandwidth must be sufficiently small to resolve the features of interest, typically requiring advanced crystal monochromators for the incident beam and high-resolution analyzers for the emitted radiation. Common geometrical arrangements for RIXS instrumentation include Johann, Johansson, and van Howe geometries, all based on the Rowland circle principle using spherically bent analyzer crystals [57].
The following diagram illustrates the fundamental quantum processes involved in the RIXS technique:
Spectral Interpretation and Applications
RIXS spectra contain rich information about the electronic structure of materials. For transition metal compounds, RIXS can resolve crystal field excitations, charge transfer energies, and multiplet structures that are often obscured in conventional XAS spectra. The presence of different manganese oxidation states (Mn²⁺, Mn³⁺, Mn⁴⁺) and associated Jahn-Teller distortions can be identified through characteristic features in the RIXS spectra, as demonstrated in studies of manganite systems like LaSr₂Mn₂O₇ and La₀.₉₅Ba₀.₀₅MnO₃ [59].
The momentum transfer dependence of RIXS features enables the mapping of dispersion relations for collective excitations, making it particularly valuable for studying quantum materials with strongly correlated electrons. This capability has established RIXS as a powerful tool for investigating high-temperature superconductors, magnetic systems, and low-dimensional quantum materials [59] [57].
Experimental Protocols and Materials
Essential Research Reagents and Materials
Table 2: Essential Research Reagents and Materials for Core-Level Spectroscopy
| Item | Function/Purpose | Technical Specifications |
|---|---|---|
| Synchrotron Beamline Access | Provides intense, tunable X-ray source | Energy range covering relevant absorption edges (0.1-100 keV) |
| High-Resolution Monochromator | Selects precise incident energies | Energy resolution ΔE/E ~10⁻⁴ or better |
| Ionization Chambers | Measures incident and transmitted beam intensities | High linearity, appropriate gas fill for energy range |
| X-Ray Fluorescence Detector | Detects emitted X-rays in fluorescence mode | Solid-state detectors (e.g., Ge, Si drift diodes) |
| Spherically Bent Analyzer Crystals | Energy analysis of emitted radiation for RIXS | Quartz, Si, Ge crystals with Rowland circle geometry |
| Sample Cells/Holders | Contains samples under controlled environments | Compatible with vacuum, various temperatures, in situ conditions |
| Reference Compounds | Energy calibration and spectral interpretation | Well-characterized materials with known oxidation states |
Standard Experimental Protocol for XAS
Sample Preparation: Prepare homogeneous samples appropriate for the detection mode. For transmission measurements, optimize sample thickness to achieve an absorbance (μd) of approximately 1. For fluorescence measurements, ensure uniform distribution of the target element. Solid powders may be pressed into pellets with boron nitride or cellulose as diluents [33].
Energy Calibration: Collect spectra of appropriate reference compounds (e.g., metal foils for K-edges) simultaneously with the sample or immediately before/after sample measurement to establish accurate energy calibration [33].
Data Collection: Acquire spectra across the relevant energy range, typically from 200 eV below to 1000 eV above the absorption edge. Multiple scans (typically 3-10) are averaged to improve signal-to-noise ratio [33].
Data Processing: Perform background subtraction, energy calibration, and normalization using specialized software (e.g., ATHENA, LARCH). For fluorescence data, apply self-absorption corrections if necessary [33].
Data Analysis: Interpret XANES region through linear combination fitting or principal component analysis. Analyze EXAFS region by fitting theoretical models to extract structural parameters [33] [56].
Advanced Protocol for RIXS Measurements
Incident Energy Selection: Perform XAS scan to identify resonant features of interest. Select incident energies corresponding to pre-edge, edge, and above-edge features for detailed RIXS maps [57].
Energy Transfer Calibration: Establish accurate energy transfer scale (ℏωin - ℏωout) by measuring elastic scattering from a non-resonant sample or using reference emission lines [57].
RIXS Map Acquisition: Collect full RIXS maps by scanning both incident energy and emitted energy across regions of interest. Acquisition times vary significantly based on cross-sections and required statistics [57].
Data Processing: Correct for instrumental background, detector efficiency, and self-absorption effects. Extract constant-incident-energy cuts or constant-energy-transfer cuts as needed for specific analyses [57].
Spectral Interpretation: Analyze RIXS features with reference to theoretical calculations, including crystal field multiplet theory, density functional theory, or exact diagonalization approaches [59] [57].
Applications in Materials and Pharmaceutical Research
Core-level spectroscopy techniques have enabled significant advances across multiple scientific disciplines. In energy conversion and storage, XAS and RIXS have been instrumental in characterizing electrode materials for batteries, fuel cells, and electrolyzers, particularly in understanding the role of transition metals and light elements in determining performance and lifespan [58]. These techniques provide direct insights into oxidation state changes during electrochemical cycling, degradation mechanisms, and the relationship between electronic structure and device performance.
In pharmaceutical research, despite currently limited application, XAS and XES show considerable promise for analyzing crystalline active pharmaceutical ingredients (APIs), drug-biomolecule interactions, and differences in drug activity. The element selectivity of these techniques enables the specific study of metal-containing drugs without matrix interference, allowing researchers to determine metal speciation, oxidation states, and coordination environments in complex biological systems [33].
The application of these techniques to complex quantum materials is exemplified by studies of manganite systems, where RIXS, XES, and XAS have revealed the presence of multiple manganese oxidation states (Mn²⁺, Mn³⁺, Mn⁴⁺) and Jahn-Teller distortions associated with overlapping between O²⁻ and unpaired electrons of manganese. These electronic structure parameters are crucial for understanding the magnetic transitions, metal-insulator transitions, and colossal magnetoresistance properties of these materials [59].
Core-level spectroscopy techniques, particularly XAS and RIXS, provide powerful and complementary approaches for probing the electronic structure of materials with element specificity and high chemical sensitivity. XAS offers direct information about unoccupied states, oxidation states, and local atomic structure, while RIXS enables the study of low-energy excitations with enhanced resolution. The ongoing development of synchrotron radiation sources, with increasing brightness and energy resolution, continues to expand the capabilities of these techniques, opening new possibilities for studying increasingly complex systems under realistic operating conditions.
For researchers in pharmaceutical development and materials science, these techniques offer unique insights that complement other analytical methods. The ability to probe specific elements in complex matrices without long-range order requirements makes XAS and RIXS particularly valuable for studying amorphous pharmaceuticals, protein-metal complexes, and operational energy storage devices. As these techniques become more accessible and their theoretical framework continues to mature, their application to challenging problems in electron energy level transitions spectroscopy is expected to grow significantly, driving advances in both fundamental understanding and technological applications.
Enhancing Sensitivity: Overcoming Challenges in Spectral Detection
In the field of electron spectroscopy and electronic structure research, the study of energy level transitions forms the cornerstone of understanding material and molecular properties. However, a significant challenge persists: the experimental detection of weak transitions. According to Fermi's golden rule, the absorption cross-section in traditional spectroscopy scales with the absolute square of the transition matrix element[cite:7]. This fundamental scaling law means that transitions with inherently small matrix elements—often due to symmetry constraints or complex multi-electron interactions—produce signals that are easily lost in noise or obscured by competing background processes[cite:7]. Such limitations hinder progress in multiple domains, from analytical biochemistry to precision physics, where these faint signatures can carry crucial information about electronic structure, bonding environments, and excited-state dynamics.
This technical guide examines a groundbreaking approach that breaks this traditional scaling law by exploiting quantum-mechanical pathway interference to enhance the spectral visibility of otherwise faint transitions. We frame this discussion within the broader context of electron energy level transitions spectroscopy, providing researchers with both the theoretical foundation and practical experimental methodologies to apply these techniques in their own investigations, particularly in fields like drug development where understanding subtle electronic interactions is paramount.
Theoretical Foundation: Beyond Fermi's Golden Rule
The Traditional Scaling Limitation
Conventional spectroscopy operates primarily in the linear regime, where the interaction of light with matter follows Fermi's golden rule. In this regime, the transition rate ( R{i,f} ) between initial state ( \Phii ) and final state ( \Phi_f ) is given by:
[ R{i,f} = \frac{2\pi}{\hbar^2} f(\omega{f,i}) | \textbf{E}0· \langle \Phif | \boldsymbol{\mu} | \Phi_i\rangle |^2 ]
where ( \langle \Phif | \boldsymbol{\mu} | \Phii\rangle ) represents the transition dipole matrix element, and ( f(\omega_{f,i}) ) is the photon source intensity at the transition frequency[cite:1]. The critical limitation is evident: the signal strength depends quadratically on the transition matrix element, causing weak transitions to manifest with disproportionately faint signals.
Pathway Interference: Breaking the Scaling Law
The breakthrough technique involves moving beyond the linear regime by introducing additional laser-coupled pathways to the same excited state. When a stronger transition pathway to a different excited state exists, and this state is coupled to the weakly-connected target state, the overall transition probability can be dramatically enhanced[cite:7].
The modified response function in the presence of intense light at different frequencies becomes:
[ \tilde{A}\propto {T}^{*}(T+T^{\prime} ) ]
where ( T ) represents the direct transition matrix element, and ( T^{\prime} ) accounts for contributions from additional pathways beyond direct single-photon excitation[cite:7]. For weak direct transitions where ( T^{\prime} ) can be much larger than ( T ), this interference effect can enhance spectral visibility by an order of magnitude or more, effectively breaking the traditional scaling law that has limited spectroscopic sensitivity for decades.
Experimental Demonstration in Helium
A recent experimental demonstration in helium atoms provides a compelling validation of this approach[cite:7]. The study targeted two specific doubly-excited states (2p3d and sp2,4−) with transition probabilities from the ground state (1s2) that are several orders of magnitude weaker than that of the strongly-coupled 2s2p state[cite:7]. These particular transitions are considered "quasi-forbidden" and represent an ideal test case due to their well-characterized weakness and the availability of a suitable coupling pathway.
Table: Transition Strengths in Helium Atomic System
| State | Transition Probability (Relative) | Classification |
|---|---|---|
| 2s2p | High | Strongly-coupled |
| 2p3d | Several orders lower | Quasi-forbidden |
| sp2,4− | Several orders lower | Quasi-forbidden |
Enhanced Transient Absorption Spectroscopy
The experimental setup employed a collinear beam-path transient absorption beamline where few-cycle visible (VIS) pulses propagated together with extreme-ultraviolet (XUV) pulses produced by high-harmonic generation in neon[cite:7]. The key enhancement mechanism involved strongly coupling the target states (2p3d and sp2,4−) to the 2s2p state using two visible photons via the intermediate 2p2 state.
Diagram: Enhanced Transition Pathway in Helium
When only the weak XUV pulse was present, the absorption spectrum showed predominant features from the strongly-coupled sp2,n+ series, with the sp2,n− series barely visible—consistent with historical synchrotron radiation measurements[cite:7]. However, with the additional VIS pulse applied, the spectral features around the weakly-coupled 2p3d and sp2,4− resonances at ~64.1 eV showed dramatic enhancement, becoming comparable in visibility to neighboring strong transitions.
Experimental Protocol & Methodology
Apparatus Configuration
- Beamline Setup: Collinear geometry for XUV and VIS pulses with variable time delay controlled by piezo-driven split mirror[cite:7]
- XUV Generation: High-harmonic generation in neon gas cell
- Pulse Characteristics:
- XUV: 0.2 fs FWHM duration, 1×10¹¹ W/cm² peak intensity, centered at 60 eV
- VIS: 700 nm central wavelength, 4 fs FWHM duration, 2×10¹² W/cm² peak intensity
- Detection: Transmitted XUV radiation dispersed by grating and detected by CCD camera
- Spectral Resolution: 40 meV near 63.7 eV[cite:7]
Critical Timing Parameters
The temporal overlap between XUV and VIS pulses proved crucial for observing enhancement effects. The spectral enhancement of weak transitions:
- Was barely visible at negative time delays (XUV arriving before VIS)
- Became pronounced in the pulse-overlap region
- Gradually faded with increasing positive time delay
- Exhibited a finite observation window linked to the ~17-fs lifetime of the 2s2p intermediate state[cite:7]
Data Processing and Analysis
The experimental optical density was calculated as:
[ {{{{\rm{OD}}}}}(\omega,\tau )=-{\log }{10}[I(\omega,\tau )/{I}{0}(\omega )] ]
where ( I(\omega, \tau) ) and ( I_0(\omega) ) represent transmitted and incident XUV spectra, respectively[cite:7]. Theoretical modeling employed large-scale numerical simulation by solving the two-electron time-dependent Schrödinger equation (TDSE) using the hyperspherical close-coupling method, with subsequent inclusion of spectral broadening and finite spectrometer resolution for direct comparison with experimental results.
The Scientist's Toolkit: Essential Research Reagents & Materials
Table: Essential Research Reagents and Materials for Enhanced Transition Spectroscopy
| Reagent/Material | Function/Significance |
|---|---|
| Helium Target Gas | Model system for method validation; provides well-characterized weak transitions |
| Neon Gas Cell | Medium for high-harmonic generation to produce XUV pulses |
| Few-Cycle VIS Laser Source | Provides intense coupling pathway for enhanced transition probability |
| Piezo-Driven Split Mirror | Enables precise temporal delay between XUV and VIS pulses |
| CCD Detection System | Captures transmitted XUV spectra with high sensitivity |
| Spectrometer Grating | Disperses XUV radiation for spectral analysis |
Complementary Techniques in Quantum-Confined Systems
Research in Ruddlesden-Popper halide perovskites (RPPs) provides additional insights into measuring and controlling excitonic properties in quantum-confined systems. These 2D solution-processed quantum wells with general formula A₂A'ₙ₋₁MₙX₃ₙ₊₁ allow systematic tuning of optoelectronic properties by varying perovskite layer thickness (n-value)[cite:4].
Magneto-optical spectroscopy under high magnetic fields (up to 60 Tesla) has enabled precise determination of exciton reduced masses and binding energies in these systems[cite:4]. The diamagnetic coefficient ( c_0 ), which increases monotonically with perovskite layer thickness from ~0.2 to ~1.1 μeV T⁻² for n=1 to 5, provides critical information about electron-hole Coulomb interactions[cite:4].
Table: Exciton Properties vs. Quantum Well Thickness in RPPs
| Layer Thickness (n) | Exciton Reduced Mass (m₀) | Binding Energy (meV) |
|---|---|---|
| 1 | 0.221 | 470 |
| 2 | 0.217 | - |
| 3 | 0.201 | - |
| 4 | 0.192 | - |
| 5 | 0.186 | 125 |
This complementary approach demonstrates how strategic selection of material systems can provide enhanced access to fundamental electronic parameters through thickness-dependent scaling laws.
The technique of enhancing weak transitions through coupled laser pathways represents a significant advancement in spectroscopic capabilities, effectively breaking the traditional scaling law that has limited sensitivity for decades. The experimental demonstration in helium, achieving an order-of-magnitude enhancement for quasi-forbidden transitions, provides a roadmap for applications across multiple disciplines.
For researchers in drug development and biochemistry, these methods offer potential solutions for detecting subtle electronic transitions in complex molecular systems where weak signals often carry critical information about molecular structure and interactions. The principles outlined—pathway interference, precise temporal control, and strategic coupling of quantum states—can be adapted to molecular spectroscopy, potentially unlocking new analytical capabilities for studying pharmaceutical compounds and biological systems.
Future developments will likely focus on extending these concepts to more complex molecular systems, optimizing enhancement factors for specific applications, and integrating these approaches with other spectroscopic techniques to provide comprehensive electronic structure characterization.
This technical guide examines the critical roles of temperature control and solvent selection in mitigating experimental noise within spectroscopic studies of electron energy level transitions. Effective management of these factors is essential for obtaining high-fidelity data, particularly in sensitive fields such as pharmaceutical development where precise characterization of molecular electronic properties directly impacts drug efficacy and safety profiling. We present a comprehensive analysis of noise mitigation protocols, supported by quantitative data and detailed methodologies for implementation in research settings.
Electronic spectroscopy probes the quantized nature of energy states by measuring the absorption and emission of light as electrons transition between energy levels. Without proper experimental controls, these measurements are susceptible to significant noise that obscures true electronic properties. Temperature fluctuations cause broadening of spectral bands and introduce vibrational hot bands, while solvent interactions shift transition energies through solute-solvent interactions, potentially masking critical electronic information [22] [60]. Within drug development, where spectroscopic characterization informs molecular design, controlling these variables ensures accurate determination of frontier molecular orbitals, charge transfer behavior, and reactive sites—properties fundamental to predicting biological activity [60].
The fundamental process involves exciting an electron from its ground state to a higher energy excited state through photon absorption. The subsequent relaxation to a lower state releases energy detectable as fluorescence. Embedded within these electronic states are vibrational levels (v=1,2,3,...) and rotational energy levels (j=1,2,3,...), all susceptible to environmental perturbations [22]. Understanding and controlling these sources of noise is therefore prerequisite to reliable spectroscopic analysis in research applications.
Theoretical Framework: Environmental Perturbation of Electronic Transitions
Solvent Effects on Electronic Energy Levels
Solvent environments significantly influence a molecule's electronic structure through several mechanisms. The integral equation formalism polarisable continuum model (IEFPCM) effectively simulates these interactions by treating the solvent as a continuous dielectric field [60]. Polar solvents stabilize excited states more effectively than ground states, leading to red-shifted absorption spectra—a phenomenon observed in 2-[(trimethylsilyl)ethynyl]thiophene (2TSET) studies across different solvent environments [60]. Specific solvent interactions include:
- Polarity Effects: Higher polarity solvents decrease energy gaps between frontier molecular orbitals (HOMO-LUMO), reducing transition energies.
- Hydrogen Bonding: Protic solvents (e.g., water, ethanol) specifically interact with electronegative atoms in solute molecules, altering electronic density distributions.
- Polarizability: Solvent electron clouds interact transiently with solute molecules, creating induced dipole moments that stabilize electronic states.
These solvent-driven perturbations necessitate careful environmental control during spectroscopic characterization of drug candidates, as electronic properties directly influence binding interactions with biological targets [60].
Temperature Effects on Spectral Resolution
Temperature directly influences spectroscopic resolution through its effect on molecular motion. Decreasing temperature reduces molecular collision frequency and population of excited vibrational states, thereby sharpening spectral features [22]. This phenomenon is particularly crucial for resolving vibrational fine structure within electronic transitions, as demonstrated by the emergence of distinct vibrational bands at cryogenic temperatures [22]. The underlying mechanisms include:
- Vibrational Hot Bands: At elevated temperatures, transitions originating from vibrationally excited ground states (v>0) create additional spectral features that complicate interpretation.
- Doppler Broadening: Thermal motion of molecules causes frequency shifts through the Doppler effect, broadening spectral lines.
- Collisional Broadening: Intermolecular collisions at higher temperatures decrease excited state lifetimes through energy transfer, widening spectral bands.
Table 1: Quantitative Effects of Temperature on Spectral Features
| Temperature Condition | Effect on Vibrational Fine Structure | Impact on Signal-to-Noise Ratio | Typical Application |
|---|---|---|---|
| Room Temperature (298K) | Broadened, averaged features | Moderate | Routine screening |
| Reduced Temperature (77K) | Enhanced resolution | High | Detailed characterization |
| Cryogenic (4-20K) | Atom-like sharp lines | Very High | High-precision studies |
Experimental Protocols for Noise Mitigation
Solvent Selection and Preparation Methodology
Protocol for Solvent-Dependent Spectroscopic Analysis
Solvent Screening:
- Select solvents spanning a range of polarities (e.g., water, ethanol, DMSO, non-polar organic solvents)
- Characterize solvent purity using chromatography or blank spectroscopic scans
- Degas solvents using freeze-pump-thaw cycles to eliminate oxygen quenching effects
Sample Preparation:
Spectroscopic Measurement:
- Utilize quartz cuvettes with 1 cm path length for UV-Vis measurements [60]
- Perform baseline correction with pure solvent reference
- Maintain constant temperature using jacketed cuvette holders during measurements
Data Analysis:
Temperature Control Methodology
Protocol for Temperature-Dependent Spectral Acquisition
Temperature Stabilization:
- Use liquid nitrogen cryostats for temperature control below ambient
- Implement heated cell holders with PID controllers for elevated temperatures
- Allow 15-20 minutes thermal equilibration after temperature changes
Measurement Parameters:
Data Correction:
- Apply temperature-dependent baseline correction
- Normalize spectra to account for density changes with temperature
- Use generalized quantum mechanical force field methods for vibrational analysis [60]
Table 2: Research Reagent Solutions for Spectroscopic Studies
| Reagent/Material | Function | Specification Guidelines |
|---|---|---|
| Spectroscopic-grade solvents | Minimize impurity interference | Low UV cutoff, fluorescence-free |
| Quartz cuvettes | UV-Vis sample containment | 1 cm path length, fused quartz [60] |
| KBr pellets | FT-IR sample preparation | 1:100 sample:KBr ratio, 10-ton pressure [60] |
| Mueller Hinton Agar | Antibacterial activity testing | Standardized growth medium for zone inhibition assays [60] |
| ND:YAG laser | Raman excitation | 1064 nm wavelength, 150-250 mW power [60] |
| IEFPCM computational model | Solvent effect simulation | Implicit solvation for DMSO, ethanol, water [60] |
Data Presentation and Analysis
Quantitative Solvent Effects on Electronic Properties
Computational and experimental studies of 2-[(trimethylsilyl)ethynyl]thiophene (2TSET) demonstrate significant solvent dependence in electronic properties. Frontier molecular orbital energies shift systematically with solvent polarity, affecting intramolecular charge transfer characteristics [60]. Time-dependent density functional theory (TD-DFT) calculations at the B3LYP/6-311+G(d,2p) level accurately predict these solvatochromic shifts when employing polarizable continuum models [60].
Table 3: Solvent Effects on 2TSET Electronic Transitions
| Solvent Environment | HOMO-LUMO Gap (eV) | Absorption Maximum (nm) | Fluorescence Shift | Computational Method |
|---|---|---|---|---|
| Gas Phase | 4.85 | 285 | Reference | DFT/B3LYP/6-311+G(d,2p) |
| Water (ε=78.36) | 4.72 | 292 +7 nm | IEFPCM Continuum Model | |
| Ethanol (ε=24.55) | 4.78 | 289 +4 nm | IEFPCM Continuum Model | |
| DMSO (ε=46.68) | 4.75 | 291 +6 nm | IEFPCM Continuum Model |
Temperature-Dependent Spectral Resolution
The emergence of vibrational fine structure at reduced temperatures provides direct evidence for temperature-mediated noise reduction. Studies demonstrate that cooling samples to cryogenic temperatures (77K) reveals vibrational progressions completely obscured at room temperature [22]. This resolution enhancement enables precise assignment of vibrational modes and validation of computational models through experimental comparison.
Visualization of Experimental Workflows
Diagram 1: Experimental workflow for noise-controlled spectroscopy
Diagram 2: Environmental effects on energy level transitions
Implementation in Pharmaceutical Research
The described methodologies find direct application in drug development pipelines. For 2TSET, molecular docking studies demonstrated strong binding affinity (-4.9 kcal/mol with 2J6M, -4.6 kcal/mol with 3ERT), highlighting its potential as an antimicrobial agent [60]. Accurate spectroscopic characterization under controlled conditions enabled precise determination of electronic properties that correlate with bioactivity. Implementation protocols include:
- ADMET Profiling: Using solvent-dependent partition coefficients predicted from spectroscopic data
- Bioactivity Scoring: Correlating HOMO-LUMO gaps with inhibitory potential
- Binding Affinity Prediction: Utilizing electrostatic potential maps from spectroscopic analysis
Zone inhibition assays conducted with 2TSET against drug-resistant superbugs (20-80 μg/mL concentrations) validated computational predictions, demonstrating the critical importance of accurate physicochemical characterization in therapeutic development [60].
Temperature and solvent effects represent significant sources of experimental noise in spectroscopic studies of electron energy level transitions. Through systematic implementation of the protocols outlined in this guide—including rigorous solvent selection, temperature control, and computational modeling—researchers can significantly enhance data quality and reliability. These noise mitigation strategies prove particularly valuable in pharmaceutical applications where precise electronic structure characterization informs drug design and optimization. Future advancements in multiscale modeling and in situ spectroscopic monitoring will further refine our ability to disentangle environmental effects from intrinsic molecular properties.
Optimizing Charge Transport and Collection in Optoelectronic Sensors
The performance of optoelectronic sensors is fundamentally governed by the efficiency of charge carrier transport and the subsequent collection of these charges at electrodes. This process is intrinsically linked to the electron energy level landscape within the active materials. Electron energy level transitions spectroscopy, which encompasses techniques like Ultraviolet Photoelectron Spectroscopy (UPS) and X-ray Photoelectron Spectroscopy (XPS), provides critical insights into these energy levels, enabling researchers to probe the electronic structure of materials with high precision [61]. Optimizing the interfaces and bulk properties based on this understanding is paramount for developing advanced sensors with high responsivity, low noise, and fast response times. This guide details the core principles, materials, characterization methodologies, and optimization strategies central to this endeavor.
Theoretical Foundations: Charge Transport and Electron Transitions
Charge Transport Mechanisms
In optoelectronic sensors, photon absorption generates excitons (bound electron-hole pairs), which must dissociate into free charges. These charges then transport through the material under the influence of built-in electric fields and concentration gradients, eventually being collected at the electrodes. The efficiency of this journey from photon to collected current defines the sensor's quantum efficiency. Charge transport can occur via band transport in crystalline inorganic semiconductors or through hopping mechanisms in disordered organic and hybrid materials. The latter is highly dependent on the molecular organization and energy level alignment at heterointerfaces.
Spectroscopy of Electron Energy Level Transitions
Electronic Spectroscopy relies on the quantized nature of energy states, where electrons are excited from a ground state to a higher energy excited state by absorbing energy [22]. The measured energies of these transitions provide a fingerprint of the material's electronic structure.
- Photoelectron Spectroscopy (PES): This technique uses ionizing radiation (UV or X-rays) to eject electrons from atomic and molecular orbitals. The measured kinetic energies of these ejected electrons allow for the direct determination of their original binding energies [61]. UPS is particularly sensitive to valence band states, while XPS probes core levels and provides chemical state information.
- Optical Emission Spectroscopy (OES): This method involves exciting an electron to a higher energy level and measuring the light emitted as it returns to a lower energy state [30]. The difference in energy levels determines the wavelength of the emitted photon.
- X-ray Fluorescence (XRF): When a core-level electron is ejected, an electron from a higher shell can fill the vacancy. The excess energy from this transition may be released as a characteristic X-ray [30].
Table 1: Electron Spectroscopy Techniques for Energy Level Analysis
| Technique | Excitation Source | Information Obtained | Key Application in Sensor Research |
|---|---|---|---|
| XPS/ESCA | X-rays | Core-level binding energy, chemical composition, empirical formula [61] | Quantifying energy level alignment at interfaces, identifying chemical degradation. |
| UPS | Ultraviolet light | Valence band structure, work function, ionization potential [61] | Measuring energy level alignment for charge injection barriers. |
| AES | Electron beam | Elemental composition, chemical environment (from Auger peaks) [61] | Surface-specific analysis of electrode materials and interfacial layers. |
| OES | Thermal/Electrical | Energy of outer-shell electron transitions [30] | Probing band gaps and defect states in semiconductor materials. |
Material Systems and Charge Transport Properties
The choice of active material is critical, as its molecular and electronic structure dictates charge transport pathways. Recent research highlights promising material classes.
π-Extended Carbo[n]Helicenes constitute a unique class of conjugated polycyclic aromatic hydrocarbons with a chiral helical structure. Their twisted topology leads to interesting electronic and chiroptical properties relevant for organic optoelectronics [62]. Computational studies using Density Functional Theory (DFT) reveal that their electronic properties are tunable with molecular size.
Table 2: Electronic Properties of π-Extended Carbo[n]Helicenes (Theoretical Data)
| Helicene | HOMO-LUMO Gap (eV) | Simulated Absorption Range (nm) | Reorganization Energy, λ (eV) |
|---|---|---|---|
| [7]Helicene | 3.83 | ~370 | λh < λe (for all) |
| [9]Helicene | 3.69 | ~390 | - |
| [11]Helicene | 3.56 | ~410 | - |
| [13]Helicene | 3.45 | ~430 | - |
| [15]Helicene | 3.37 | ~450 | - |
| [17]Helicene | 3.32 | ~465 | - |
Key findings from these studies show that the band gap of Carbo[n]Helicenes decreases as the helicity (number of rings) increases, which enhances the optoelectronic character for charge transfer in the materials [62]. Furthermore, the fact that the hole reorganization energy (λh) is lower than the electron reorganization energy (λe) suggests these materials are better suited for hole transport, a crucial consideration for device design.
Experimental Protocols for Characterization and Optimization
Protocol 1: Computational Screening of Molecular Materials
This protocol uses DFT to predict a material's charge transport properties before synthesis.
- Geometry Optimization: Perform molecular structure optimization using a functional like B3LYP and a basis set such as 6-311G(d,p) in a computational package (e.g., Gaussian). Confirm the structure is at a true energy minimum by verifying no imaginary frequencies are present [62].
- Electronic Property Calculation:
- Calculate the Frontier Molecular Orbitals (FMOs) - the Highest Occupied Molecular Orbital (HOMO) and Lowest Unoccupied Molecular Orbital (LUMO). The energy difference is the HOMO-LUMO gap.
- Compute the Ionization Potential (I.P.) and Electron Affinity (E.A.), which relate to the HOMO and LUMO energies, respectively.
- Evaluate the reorganization energy (λ) for both holes (λh) and electrons (λe). A lower reorganization energy indicates a more favorable charge transport property [62].
- Optical Property Simulation: Use Time-Dependent DFT (TD-DFT) to simulate the absorption spectrum and derive key nonlinear optical (NLO) parameters, such as hyperpolarizability [62].
Protocol 2: Characterizing Charge Transport in Single-Molecule Junctions
Single-molecule devices are powerful platforms for directly studying charge transport mechanisms, free from the averaging effects of bulk materials [63].
- Device Fabrication: Create nanogap electrodes (e.g., using mechanical break junctions or electromigration). Molecules of interest are then anchored to these electrodes via specific chemical link groups (e.g., thiols, amines).
- Photo-Modulation: Illuminate the molecular junction with a tunable light source. The photon energy can be adjusted to match specific electronic transitions within the molecule, thereby modulating the charge transport by populating excited states [63].
- Current-Voltage (I-V) Measurement: Record the electrical current through the molecule as a function of applied bias voltage, both in the dark and under illumination. Analyzing these curves provides information on the charge transport mechanism (e.g., coherent tunneling vs. hopping) and the photo-induced current gain.
Experimental Workflow Diagram
The following diagram illustrates a generalized workflow for developing and characterizing materials for optoelectronic sensors, integrating the protocols above.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Materials and Their Functions in Sensor Research
| Material/Reagent | Function in Research & Development |
|---|---|
| π-Extended Carbo[n]Helicenes | Active layer material with tunable band gap and intrinsic chirality for specialized optoelectronic applications [62]. |
| Electron Transport Layers (e.g., ZnO, TiO₂) | Facilitates selective collection of electrons and blocks holes, reducing charge recombination at the anode. |
| Hole Transport Layers (e.g., PEDOT:PSS, Spiro-OMeTAD) | Facilitates selective collection of holes and blocks electrons, reducing charge recombination at the cathode. |
| Chemical Linkers (e.g., Thiols, Amines) | Anchor molecules to metal electrodes in single-molecule junction studies for reliable electrical contact [63]. |
| Monochromated Light Source | Provides precise photon energies for photo-modulation experiments and external quantum efficiency (EQE) measurements [63]. |
Optimizing charge transport and collection is a multi-faceted challenge that requires a deep understanding of electron energy levels and their influence on material properties. By leveraging spectroscopic techniques like XPS and UPS to characterize energy level alignment and employing computational and experimental protocols to screen and optimize materials, researchers can systematically engineer high-performance optoelectronic sensors. The emerging understanding of structure-property relationships in novel materials like helicenes, combined with insights from single-molecule studies, provides a robust pathway for the development of next-generation sensing technologies.
Strategic Use of Buffer Layers to Reduce Leakage Current and Enhance Stability
The performance and longevity of modern electronic and optoelectronic devices are critically dependent on the management of charge carriers and the structural integrity of their layered architectures. Buffer layers, strategically inserted between the active and electrode layers, have emerged as a cornerstone technology for achieving this stability. Their primary function is to engineer the electronic energy landscape at interfaces, which directly governs the flow of charge. A key manifestation of this control is the significant reduction of leakage current—an undesirable flow of charge in the "off" state of a device that increases power consumption, generates noise, and diminishes detection sensitivity. The strategic use of these layers is not merely an empirical optimization but can be fundamentally understood and designed through the lens of electron energy level transitions spectroscopy, which provides a detailed map of the energy states involved in charge transport and recombination processes.
The efficacy of a buffer layer is determined by its ability to modify the energy level alignment at an interface. Mismatched electron energy levels between adjacent layers can create charge traps or low-resistance pathways, leading to high leakage currents. Spectroscopy research techniques, such as ultraviolet photoelectron spectroscopy (UPS) and inverse photoemission spectroscopy (IPES), allow scientists to directly measure the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) levels, as well as work functions. By providing a precise picture of these energy levels, spectroscopy guides the selection or design of buffer layer materials that create favorable energy step gradients or block charge carriers from leaking, thereby enhancing device performance and operational stability.
Core Principles: Leakage Current and Energy Level Transitions
The Microscopic Origin of Leakage Current
Leakage current is a quantum mechanical phenomenon rooted in the behavior of electrons at interfaces. At the microscopic level, it can arise from several mechanisms:
- Thermionic Emission: Charge carriers acquire enough thermal energy to overcome a potential energy barrier at an interface.
- Fowler-Nordheim Tunneling: Electrons quantum-mechanically tunnel through a triangular potential barrier under a high electric field.
- Direct Tunneling: Electrons tunnel through a thin, finite potential barrier.
- Charge Trap-Assisted Tunneling: Defects or impurities within the band gap create intermediate energy states that "step" electrons through the barrier, significantly increasing leakage.
The insertion of a tailored buffer layer directly counteracts these mechanisms by modifying the potential energy profile of the device. It can widen the tunneling barrier, increase the height of the energy barrier for thermionic emission, or passivate interfacial defects to eliminate trap states.
Spectroscopy of Electron Transitions
Electronic Spectroscopy relies on the quantized nature of energy states. When a material absorbs a photon with energy matching the gap between two electronic states, an electron can be excited from a lower energy level to a higher one. The resulting absorbance spectrum provides a fingerprint of the allowed electronic transitions within a material [22].
This is directly applicable to analyzing buffer layers and their interfaces. By studying the spectroscopic signatures, researchers can determine:
- Energy Level Offsets: The difference between the HOMO levels of the donor and acceptor materials, or the LUMO levels, which dictates the driving force for charge separation.
- Gap States: The presence of sub-bandgap states, observable in the spectrum, indicates trap states that can facilitate leakage via trap-assisted tunneling.
- Molecular Orbitals Involved: Spectroscopy can distinguish between different types of transitions, such as π-π* transitions in organic semiconductors or Metal to Ligand Charge Transfer (MLCT) in coordination complexes, informing material choice [22].
Understanding these transitions is crucial for predicting and verifying how a buffer layer will behave. For instance, a buffer layer that introduces a large energy barrier, as confirmed by spectroscopy, will effectively block electrons and reduce leakage current.
Quantitative Data from Experimental Studies
The following table summarizes key performance enhancements achieved by implementing buffer layers in two distinct device types, as revealed by recent research.
Table 1: Performance Enhancement with Buffer Layers in Different Devices
| Device Type | Buffer Layer Material & Structure | Key Function of Buffer Layer | Impact on Leakage Current | Resultant Key Performance Metric | Ref. |
|---|---|---|---|---|---|
| GaN-based High-Electron Mobility Transistor (HEMT) | Carbon-doped GaN Superlattice | Suppresses leakage pathways in the buffer [64]. | Significant reduction | Improved transistor performance and reliability [64]. | |
| Organic Photodetector (OPD) | (PEI/PAA)₂ Polyelectrolyte Multilayer | p-type buffer layer enabling light-assisted tunneling [65]. | Critical suppression of reverse-biased leakage current | Specific Detectivity: 3.11 × 10¹² Jones (at -1 V, 525 nm) [65]. |
Experimental Protocols for Buffer Layer Implementation
Protocol A: Spin-Assisted Layer-by-Layer (LbL) Assembly of Polyelectrolyte Buffer Layers
This protocol details the methodology for creating the PEI/PAA buffer layer used in the OPD study cited in Table 1 [65].
4.1.1 Research Reagent Solutions
Table 2: Essential Materials for Polyelectrolyte Buffer Layer Fabrication
| Research Reagent | Function / Explanation |
|---|---|
| Polyethyleneimine (PEI) | A polycationic polymer; serves as the positively charged component in the multilayer structure, adhering to negatively charged surfaces like ITO. |
| Polyacrylic acid (PAA) | A polyanionic polymer; serves as the negatively charged component, forming ionic bonds with PEI to build the multilayer film. |
| ITO-coated Glass Substrate | The transparent anode. The buffer layer is deposited directly onto this surface. |
| Solvents (e.g., deionized water, adjusted to specific pH) | The medium for dissolving PEI and PAA. The pH is critical as it controls the ionization degree and conformation of the polyelectrolytes, affecting film thickness and roughness. |
4.1.2 Step-by-Step Methodology
- Substrate Preparation: Clean the ITO-coated glass substrates thoroughly with solvents (e.g., acetone, isopropanol) in an ultrasonic bath, followed by oxygen plasma treatment to create a hydrophilic, negatively charged surface.
- Polyelectrolyte Solution Preparation: Prepare aqueous solutions of PEI and PAA (typical concentration range: 0.1-1.0 mg/mL). Adjust the pH of each solution using HCl or NaOH to optimize the charge density and polymer chain conformation (e.g., pH 4.5 for PAA, pH 7-10 for PEI).
- Layer-by-Layer Deposition (for a 2-bilayer (PEI/PAA)₂ film):
- a. First PEI Layer: Deposit the PEI solution onto the spinning ITO substrate (spin-coating). The substrate's negative charge attracts the positive PEI chains. Spin parameters: 3000 rpm for 20-30 seconds.
- b. Rinsing: Rinse the substrate with a stream of deionized water while spinning to remove loosely bound polymer chains, leaving only a monomolecular adsorbed layer.
- c. First PAA Layer: Deposit the PAA solution onto the spinning substrate. The negative PAA chains adsorb onto the positive PEI underlayer.
- d. Rinsing: Repeat the rinsing step to remove excess PAA.
- e. Repetition: Repeat steps (a) through (d) to build the desired number of bilayers. For the cited study, the cycle was repeated twice to create a 2-bilayer structure, denoted as (PEI/PAA)₂ [65].
- Drying: Gently dry the coated substrate under a stream of nitrogen or on a hotplate at low temperature (e.g., 50-70°C) to remove residual water before depositing subsequent device layers.
Protocol B: Superlattice Buffer Layer Growth via Epitaxial Techniques
This protocol outlines the general process for growing the carbon-doped GaN superlattice buffer layer for HEMT devices [64].
4.2.1 Research Reagent Solutions
Table 3: Essential Materials for GaN Superlattice Growth
| Research Reagent | Function / Explanation |
|---|---|
| Gallium (Ga) Source | Metal-organic precursor (e.g., Trimethylgallium - TMGa) providing Ga atoms for crystal growth. |
| Nitrogen (N) Source | Precursor (e.g., Ammonia - NH₃) providing N atoms for crystal growth. |
| Carbon (C) Doping Source | Intentional dopant introduced via a precursor like Carbon Tetrachloride (CCl₄) to create a superlattice that traps dislocations and compensates for unintentional background conductivity. |
| Single-Crystal Substrate (e.g., Sapphire, SiC) | The base upon which the crystalline GaN layers are epitaxially grown. |
4.2.2 Step-by-Step Methodology
- Substrate Loading and Preparation: Load the substrate into a metal-organic chemical vapor deposition (MOCVD) or molecular beam epitaxy (MBE) reactor chamber. Heat the substrate to a high temperature (typically >1000°C in MOCVD) under a hydrogen or nitrogen atmosphere to remove surface contaminants.
- Nucleation Layer Growth: Grow a thin, low-temperature GaN or AlN nucleation layer to initiate crystal growth and manage the lattice mismatch between the substrate and the subsequent GaN layers.
- Superlattice Growth: Grow the carbon-doped GaN superlattice buffer layer by cyclically performing the following:
- a. GaN Layer Growth: Introduce the Ga and N sources to grow a layer of undoped or lightly doped GaN of a specific thickness (a few nanometers).
- b. Carbon-Doped GaN Layer Growth: Introduce the carbon doping source along with the Ga and N sources to grow a layer of carbon-doped GaN. The carbon atoms incorporate into the crystal lattice, creating a layer with high resistivity.
- c. Cycle Repetition: Repeat steps (a) and (b) numerous times (dozens to hundreds) to build a thick buffer layer with periodic vertical composition variation (the superlattice) [64].
- Device Layer Growth: After the superlattice buffer is complete, continue with the growth of the undoped GaN channel layer and the AlGaN barrier layer to form the final HEMT structure.
Visualization of Buffer Layer Mechanisms and Workflows
Energy Level Transition Diagram with Buffer Layer
The following diagram illustrates how a buffer layer modifies the electron energy level alignment at an interface to suppress leakage current, a concept central to spectroscopy-informed research.
Experimental Workflow for OPD Fabrication
This workflow diagrams the step-by-step experimental process for fabricating an organic photodetector with a polyelectrolyte buffer layer, as detailed in Protocol A.
The strategic implementation of buffer layers, guided by precise spectroscopy research into electron energy level transitions, is a powerful and indispensable approach for controlling leakage current and ensuring the stability of advanced electronic devices. As the data and protocols herein demonstrate, both organic polyelectrolyte multilayers and inorganic doped superlattices can profoundly enhance key performance metrics like specific detectivity and operational reliability. The continued development of novel buffer layer materials and deposition techniques, coupled with ever-more-detailed spectroscopic analysis of interfacial electronic structures, promises to further unlock the performance potential of next-generation electronics, from flexible photodetectors to high-power transistors.
Leveraging Machine Learning for Automated Spectral Processing and Analysis
The advent of machine learning (ML) is fundamentally transforming spectral processing and analysis, enabling unprecedented precision in the study of electron energy-level transitions. Spectroscopy techniques, while indispensable for material characterization, often produce weak signals that are highly prone to interference from environmental noise, instrumental artifacts, and scattering effects, which can significantly degrade measurement accuracy and impair subsequent analysis [66]. This technical guide details how ML methodologies not only automate complex preprocessing and calibration tasks but also significantly enhance spectral resolution and signal-to-noise ratio. A focus on electron energy-loss spectroscopy (EELS) and orbital angular momentum (OAM) measurements demonstrates the profound impact of these tools, which are critical for advanced applications in pharmaceutical development, materials science, and quantum research.
Spectroscopic analysis of electron energy-level transitions provides a window into the fundamental electronic structure of materials. Techniques like electron energy-loss spectroscopy (EELS) in transmission electron microscopes allow the investigation of composition, valence state, and electronic level occupation at the atomic scale [67]. However, the inherent stochasticity of photon sources, such as free-electron lasers (FELs), and the pervasive challenge of weak, noisy signals complicate the extraction of clear, quantifiable insights [66] [68].
The field is undergoing a transformative shift driven by key innovations: context-aware adaptive processing, physics-constrained data fusion, and intelligent spectral enhancement [66]. Machine learning stands at the core of this shift, offering a pathway to automate diagnostic procedures, improve control, and enhance the quality of spectral diagnostics [68]. By leveraging underlying data structures and physical principles, ML models can suppress noise, correct artifacts, and resolve features beyond the intrinsic limitations of conventional diagnostic devices, thereby enabling more reliable, automated, and insightful spectroscopic research.
Machine Learning Approaches to Spectral Enhancement
The Virtual Spectrometer: A Case Study in Data Fusion
A pioneering application of ML for spectral enhancement is the "virtual spectrometer" (VS) deployed at the European XFEL. This tool addresses a critical challenge: obtaining high-resolution, pulse-resolved x-ray spectra non-invasively [68].
The core innovation lies in fusing data from multiple instruments. A grating-based spectrometer (GS) provides high-resolution spectra but is invasive and disrupts the beam. In contrast, a gas-based photo-electron spectrometer (PES) operates non-invasively at a high repetition rate but has a more complex calibration, lower signal-to-noise ratio, and lower resolution [68]. The VS uses ML to create a map from the lower-quality PES data to the high-quality GS data, effectively bestowing the PES with the resolution and calibration of the GS.
Experimental Protocol for the Virtual Spectrometer:
- Synchronous Data Collection: The X-ray gas monitor (XGM), PES, and GS collect data synchronously for an initial training period (approximately 20 minutes, capturing ~12,000 spectra) [68].
- Data Preprocessing via PCA:
- A region-of-interest is identified for all 16 PES sub-detectors.
- Principal Component Analysis (PCA) is applied to the photo-electron spectra and the pulse energy from the XGM. This step filters out a large amount of noise while preserving key features, achieving up to a nine-fold data reduction [68].
- PCA is also applied to the GS data.
- Model Training and Mapping: A fit is performed using Automatic Relevance Determination (ARD) to map the principal components from the PES to those of the GS. ARD provides robustness by setting weights with large uncertainty to zero [68].
- Inference and Quality Control: After training, the GS is removed. The VS uses only PES data and XGM pulse energies to generate high-resolution, pulse-resolved spectra. The system continuously monitors input data for drifts and provides uncertainty estimates to ensure reliability during quasi-real-time operation [68].
The following workflow diagram illustrates this integrated process:
ML-Enhanced Orbital Angular Momentum (OAM) Spectroscopy
In EELS, separating superimposed spectral features is a major challenge. For example, in hexagonal boron nitride (h-BN), the π* and σ* antibonding transitions at the boron K-edge are separated by only ~6 eV and are superimposed in a standard EELS spectrum [67]. An innovative ML-assisted approach combines EELS with an orbital angular momentum (OAM) sorter.
This technique, OAM-EELS, leverages the fact that electronic transitions are governed by selection rules involving changes in magnetic quantum number (m). The π* transition corresponds to Δm = 0, while the σ* transition corresponds to Δm = ±1 [67]. The OAM sorter acts as an electron optical device that disperses the scattered electrons based on their OAM (ℓ), physically separating these transitions.
Experimental Protocol for OAM-EELS:
- Data Acquisition: An OAM sorter is placed in the post-specimen section of a TEM. The OAM-dispersed beam is then energy-dispersed, allowing simultaneous recording of OAM and electron energy-loss in a single measurement [67].
- Point Spread Function (PSF) Characterization: A zero-loss OAM-EELS spectrum is recorded in vacuum to define the PSF of the OAM sorter, which characterizes the device's experimental resolution [67].
- Model-Based Fitting: A physical model of the inelastic scattering process is constructed, accounting for probe size, scattering localization, and probe position uncertainty. This model is fitted to the experimental OAM profile of the π* transition (expected to be m=0) [67].
- Spectral Separation: The simulated OAM profiles Γm(ℓ) for different m values are fitted to the full experimental data I(E, ℓ) to obtain a set of coefficients cm(E). This fit effectively separates the EELS spectrum into its constituent m=0 (π) and *m=±1 (σ) components according to the equation: I(E, ℓ) = Σm*cm(E)Γm(ℓ) [67].
The schematic below outlines the experimental setup and logical flow for this technique:
Quantitative Performance of ML Techniques
The application of machine learning delivers quantifiable improvements in spectral analysis. The table below summarizes key performance metrics from the cited studies.
Table 1: Quantitative Performance Metrics of ML-Enhanced Spectral Techniques
| ML Technique | Application Context | Key Performance Improvement | Reported Metrics |
|---|---|---|---|
| Virtual Spectrometer (VS) [68] | Non-invasive spectral diagnostics at European XFEL | Enhanced resolution and signal-to-noise ratio (SNR) of the Photo-electron Spectrometer (PES) | Resolution improvement of up to 40%; compatible with quasi-real-time monitoring and pulse-resolved analysis at 4.5 MHz. |
| Model-Based Fitting for OAM-EELS [67] | Separation of π* and σ* transitions at B K-edge in h-BN | Quantitative separation of overlapping spectral features | Achieved quantized OAM profiles; estimated cross-talk between channels of approximately 11%; good agreement with ab initio DFT calculations. |
| Advanced Spectral Preprocessing [66] | General spectral preprocessing for material characterization | Improved detection sensitivity and classification accuracy | Enabled detection sensitivity achieving sub-ppm levels while maintaining >99% classification accuracy. |
Essential Research Reagent Solutions
The implementation of these advanced spectroscopic and machine learning methods relies on a suite of sophisticated hardware and software tools.
Table 2: Essential Materials and Tools for ML-Enhanced Spectral Analysis
| Item Name | Function / Application |
|---|---|
| Grating Spectrometer (GS) [68] | Provides high-resolution, calibrated reference spectra for training ML models like the Virtual Spectrometer. |
| Photo-Electron Spectrometer (PES) [68] | A non-invasive diagnostic tool that provides high-repetition-rate, pulse-resolved spectral data for real-time analysis. |
| Orbital Angular Momentum (OAM) Sorter [67] | An electron optical device that disperses scattered electrons based on their orbital angular momentum, enabling the separation of transitions by their magnetic quantum number. |
| Principal Component Analysis (PCA) [68] | A dimensionality reduction technique used to denoise spectral data and identify the most relevant components before model training. |
| Automatic Relevance Determination (ARD) [68] | A robust regression method used to map data from a low-resolution instrument to a high-resolution one by pruning uncertain weights. |
| Model-Based Fitting Algorithm [67] | Uses physical models of electron scattering to deconvolve and interpret complex OAM-EELS data, separating superimposed spectral features. |
Machine learning has moved beyond a supplementary role to become a central pillar in modern spectral processing and analysis. By embracing techniques like data fusion, model-based fitting, and intelligent denoising, researchers can overcome the traditional limitations of spectroscopic instruments. The ability to automate complex calibrations, resolve superimposed spectral features with high fidelity, and provide reliable, pulse-resolved insights in real-time is accelerating the pace of discovery in fields ranging from drug development to fundamental materials science. As these ML methodologies continue to evolve, they will unlock even deeper insights into the quantum mechanical interactions that govern material behavior.
Ensuring Accuracy: Data Validation and Technique Selection
Validating Transitions and Empirical Energy Levels with Protocols like MARVEL
This guide details the MARVEL (Measured Active Rotational-Vibrational Energy Levels) protocol, a computational methodology for deriving accurate, empirical rovibrational energy levels from experimental transition data. The MARVEL algorithm constructs a self-consistent network of energy levels by analyzing all available experimental transitions, providing a validated dataset crucial for spectroscopic databases, atmospheric models, and fundamental molecular studies. This paper outlines the core principles, detailed methodologies, and practical applications of the MARVEL protocol, contextualized within electron energy level transitions spectroscopy research.
In molecular spectroscopy, the direct observation of a molecule's energy structure is achieved through its transition spectrum. When a molecule moves from a higher energy level to a lower one, it emits radiation; when it moves from a lower to a higher level, it absorbs radiation. The energy of this radiation corresponds directly to the difference between the two levels [69]. However, experimental measurements yield transitions (differences between levels), not the energy levels themselves. The primary challenge is to invert a large set of often redundant, and sometimes conflicting, transition measurements to determine the best set of underlying energy levels. This process, known as energy level validation, is fundamental to creating reliable spectroscopic databases. The MARVEL algorithm addresses this challenge by treating all experimental transitions as a network, using a weighted least-squares approach to determine the most self-consistent set of energy levels, complete with uncertainty quantification [70].
The MARVEL Protocol: A Detailed Workflow
The MARVEL protocol is a multi-stage process that transforms raw experimental data into a validated set of empirical energy levels. The workflow is designed to be rigorous, automated, and capable of handling large-scale data.
Core Principles
- Input and Output: The algorithm takes as input a set of experimental transitions, each characterized by its frequency (wavenumber in cm⁻¹), uncertainty, and quantum number assignments for the upper and lower states involved. The output is a set of empirical energy levels for each observed rovibrational state, each with a statistically derived uncertainty [70].
- Self-Consistency: The fundamental requirement is that for any closed loop of transitions (e.g., Level A → Level B, Level B → Level C, and Level C → Level A), the sum of the transition energies must be zero. MARVEL uses this principle to identify and rectify inconsistencies in the experimental data.
- Uncertainty Propagation: Each input transition's reported uncertainty is used to weight its influence on the calculated energy levels. More precise measurements have a greater impact on the final result.
Step-by-Step Methodological Breakdown
The following diagram illustrates the logical workflow of the MARVEL analysis, from data compilation to the final validated dataset.
Phase 1: Data Compilation and Curation The initial phase involves gathering all accessible experimental transition data from the literature. For a comprehensive study on sulfur monoxide (SO), this involved compiling 50,106 experimental transitions from 29 distinct experimental sources, which were then distilled into 48,972 non-redundant transitions [70]. Each data point must be accompanied by:
- Wavenumber ((\nu)): The precise frequency of the transition in cm⁻¹.
- Uncertainty ((\delta\nu)): The experimental uncertainty associated with the measurement.
- Quantum Number Assignment: Complete rovibrational and electronic state assignments for both the upper and lower states involved in the transition (e.g., (J''), (v''), electronic state' for the lower level).
Phase 2: Transition Network Construction The compiled, non-redundant transitions are used to construct a complex network. In this network:
- Nodes represent the distinct empirical rovibrational energy levels.
- Edges represent the measured transitions between these levels.
The connectivity of this network is critical. A well-connected network, where many levels are linked by multiple pathways, leads to a more robust and precise determination of energy levels.
Phase 3: Least-Squares Inversion and Uncertainty Analysis The core computational step involves solving the system of equations where each transition is expressed as the difference between two energy levels: (\nu{ij} = Ej - Ei). This overdetermined system is solved using a weighted least-squares algorithm, where the weight for each transition is typically the inverse square of its uncertainty ((1/(\delta\nu)^2)). This process yields the set of energy levels (Ei) that best fits all the experimental data. The algorithm also propagates the experimental uncertainties to compute a rigorous uncertainty for each derived energy level.
Phase 4: Validation, Refinement, and Output The final set of energy levels is scrutinized for outliers and physically implausible results. Discrepancies may lead to the re-examination of quantum number assignments or the exclusion of erroneous data. The output is a self-consistent network of empirical energy levels, which can be used to validate and refine ab initio spectroscopic models [70].
Case Study: The SOLIS Line List for Sulphur Monoxide (SO)
The recent creation of the SOLIS line list for SO exemplifies the application of the MARVEL protocol.
Experimental Data and MARVEL Analysis
The analysis compiled a massive dataset of experimental transitions, leading to a comprehensive and self-consistent set of energy levels. The table below summarizes the quantitative outcomes of the MARVEL analysis for SO.
Table 1: Summary of the MARVEL Analysis for SO (Sulphur Monoxide)
| Parameter | Value | Description |
|---|---|---|
| Total Experimental Transitions | 50,106 | Gathered from 29 sources [70] |
| Non-Redundant Transitions | 48,972 | After removing duplicate measurements [70] |
| Empirical Rovibronic Energy Levels | 8,558 | Self-consistent levels in the MARVEL network [70] |
| Covered Vibrational Quantum No. ((v)) | (\leq 30) | High vibrational excitations included [70] |
| Covered Rotational Quantum No. ((J)) | (\leq 69) | High rotational excitations included [70] |
| Maximum Energy | 52,350.40 cm⁻¹ | Energy coverage up to the UV region [70] |
Impact on Spectroscopic Modeling
The MARVEL-derived energy levels for SO were used to refine a theoretical spectroscopic model computed using the variational code Duo. The empirical levels served as a benchmark to adjust ab initio potential energy curves and coupling curves, leading to the production of the accurate SOLIS IR/Vis line list. This semi-empirical line list includes highly accurate transition probabilities (Einstein (A) coefficients) for environments ranging from exoplanetary atmospheres to industrial combustion processes [70].
Successful implementation of the MARVEL protocol and related spectroscopic research relies on a suite of computational and analytical tools.
Table 2: Key Research Reagent Solutions for Spectroscopic Validation
| Tool/Resource | Type | Primary Function |
|---|---|---|
| MARVEL Algorithm | Software Protocol | The core algorithm for deriving self-consistent empirical energy levels from experimental transitions [70]. |
| Duo | Variational Code | A general-purpose code for solving the rovibronic Schrödinger equation for diatomics, used to create line lists from a refined spectroscopic model [70]. |
| ExoMol Database | Data Repository | A repository for molecular line lists, such as the SOLIS list for SO, which are critical for atmospheric modeling [70]. |
| High-Resolution Spectrometer | Experimental Instrument | Used to obtain the primary experimental transition data (e.g., frequencies, intensities) required as input for MARVEL. |
| Einstein A Coefficients | Theoretical/Experimental Metric | Transition probabilities that provide benchmarks for intensity calculations in the spectroscopic model, often informed by experimental lifetime measurements [70]. |
Advanced Considerations and Future Directions
While the MARVEL protocol is powerful, several advanced considerations are necessary for its effective application.
- Handling Perturbations and Missing States: A notable challenge arises when observed transitions do not connect to certain perturbing states. In the SO study, the (B\,{}^{3}\Sigma^{-}) ((v=0)) state lacked connecting observations, preventing its full empirical refinement. In such cases, the theoretical model for these states must be left for future work when data becomes available [70].
- Uncertainty and Weighting Strategies: The choice of weighting functions (e.g., inverse square of uncertainty) can impact the results. Robust statistical methods are sometimes needed to handle outliers or to adjust uncertainties for systematic errors in certain datasets.
- Automated Assignment and Validation: For large datasets, automated procedures for quantum number assignment and validation are increasingly important. Machine learning approaches are being explored to assist in this labor-intensive process.
The MARVEL protocol represents a gold standard for validating transitions and deriving empirical energy levels in molecular spectroscopy. By constructing a self-consistent network from all available experimental data, it produces a reliable foundational dataset. This dataset is indispensable for testing and refining theoretical models, which in turn generate the accurate line lists required to interpret observational data from telescopes and planetary probes. As spectroscopic capabilities advance, the role of rigorous validation protocols like MARVEL will only grow in importance for fields ranging from astrochemistry to drug development where molecular identification is key.
Comparative Analysis of Spectroscopic Databases (HITRAN, CDSD, ExoMol)
Spectroscopic databases serve as critical repositories of molecular parameters essential for interpreting light-matter interactions across scientific and industrial domains. This technical analysis provides a comprehensive comparison of three premier spectroscopic databases—HITRAN, CDSD, and ExoMol—evaluating their respective architectures, applications, and performance characteristics. While HITRAN offers extensive multi-molecular coverage optimized for Earth atmospheric science, CDSD delivers specialized high-accuracy carbon dioxide parameters, and ExoMol provides comprehensive high-temperature data for exoplanetary research. Understanding the comparative strengths and limitations of these databases enables researchers to select appropriate resources for specific spectroscopic applications, from climate science and planetary studies to combustion analysis and astrophysical investigations.
Molecular spectroscopic databases compile fundamental parameters that characterize how molecules absorb and emit electromagnetic radiation through electron energy level transitions. These databases provide the essential foundation for simulating and analyzing the transmission and emission of light in gaseous media, with critical applications spanning atmospheric science, astrophysics, combustion analysis, and climate modeling [71]. The accuracy and completeness of these databases directly impact the reliability of remote sensing data interpretation, climate projections, and atmospheric characterization of celestial bodies.
Spectroscopic databases typically contain parameters including line positions (transition frequencies between energy levels), line intensities, pressure-broadening coefficients, temperature dependence parameters, and lower state energies. These parameters enable researchers to calculate absorption spectra, model radiative transfer, and retrieve atmospheric compositions from observed spectra. The three databases examined in this whitepaper—HITRAN, CDSD, and ExoMol—represent complementary approaches to addressing these needs across different temperature regimes, spectral regions, and application domains, all fundamentally rooted in the quantum mechanical principles governing electron transitions between discrete molecular energy levels.
Database-Specific Architectures and Methodologies
HITRAN Database
HITRAN (High-Resolution Transmission Molecular Absorption Database) is a comprehensive compilation of spectroscopic parameters that serves as the international standard for atmospheric radiative transfer calculations. Originally developed by the US Air Force Cambridge Research Laboratories in the 1960s, HITRAN has evolved into a multi-faceted resource maintained at the Center for Astrophysics | Harvard & Smithsonian [71]. The database employs a rigorous evaluation process, incorporating both experimental and theoretical data from a worldwide network of contributors, with validation against accurate laboratory measurements wherever possible [71].
The HITRAN architecture encompasses several distinct data modalities. The core line-by-line transition database contains parameters for millions of individual spectral lines across multiple molecules and isotopologues. Additionally, HITRAN provides absorption cross-sections for heavy polyatomic molecules where line-by-line parameters are incomplete, collision-induced absorption (CIA) data for transient dipoles formed during molecular collisions, aerosol refractive indices, and HITEMP for high-temperature applications [71]. The upcoming HITRAN2024 edition represents a substantial expansion, featuring 61 molecules in its line-by-line section and over 260 new molecules in its cross-section database, significantly enhancing its capabilities for planetary and exoplanetary studies [72].
CDSD (Carbon Dioxide Spectroscopic Databank)
CDSD is a specialized spectroscopic databank focused exclusively on carbon dioxide (CO₂) and its isotopologues. Developed by the Laboratory of Theoretical Spectroscopy at the Institute of Atmospheric Optics in Tomsk, Russia, CDSD employs the method of effective operators to generate its line parameters [73] [74]. This methodology is based on global fittings of spectroscopic parameters (effective Hamiltonians and effective dipole moment operators) to observed data collected from comprehensive literature reviews [75].
The CDSD databank is particularly notable for its systematic replacement of calculated line positions with experimental values where possible, using the differences between experimental term values derived from recalibrated observed line positions with the help of the combination Ritz principle [73]. Recent versions of CDSD have addressed previous limitations, including missing bands of the main isotopologue around 10,000 cm⁻¹ and 12,000 cm⁻¹, and incorrect intensity calculations for some asymmetric isotopologues [75]. The database includes statistically justified confidence intervals for each line position and intensity, providing users with uncertainty quantification for their analyses [73].
ExoMol Database
The ExoMol database was specifically designed to meet the needs of the exoplanet research community, providing molecular line lists for modeling hot atmospheres that are not adequately covered by terrestrial atmospheric databases. The project employs a variational approach, using ab initio quantum mechanical calculations to generate comprehensive line lists, with empirical adjustments where possible to improve accuracy [76]. Unlike HITRAN and CDSD, ExoMol prioritizes completeness for high-temperature applications, making it particularly valuable for studying exoplanets, brown dwarfs, and cool stars.
A key innovation in the ExoMol methodology is the systematic use of the MARVEL (Measured Active Rotation-Vibration Energy Levels) procedure to improve the accuracy of energy levels and transition frequencies [76]. This approach leverages experimental data to replace calculated energy levels with empirically determined ones, resulting in a significant enhancement of positional accuracy. The recently introduced ExoMolHR database provides access to the high-accuracy subset of the full ExoMol database, currently containing over 24 million spectral lines across 33 molecules and 58 isotopologues, making the strongest and most accurate transitions readily accessible for spectral assignment and analysis [76].
Comparative Analysis and Performance Metrics
Table 1: Core Database Characteristics and Applications
| Parameter | HITRAN | CDSD | ExoMol |
|---|---|---|---|
| Primary Focus | Earth atmospheric transmission & radiance | Carbon dioxide spectroscopy | High-temperature exoplanet atmospheres |
| Temperature Regime | Primarily 296 K (room temperature) | 296 K (atmospheric version) | High temperatures (up to 4000 K) |
| Molecular Coverage | 61 molecules (HITRAN2024), 144 isotopologues [72] | CO₂ and its isotopologues (8-12 species) [73] [74] | 91 molecules, 224 isotopologues (ExoMol 2024) [76] |
| Spectral Range | Microwave through ultraviolet | 405-12,784 cm⁻¹ [73] | Varies by molecule, typically IR to visible |
| Intensity Cutoff | Varies by molecule | 10⁻²⁸ to 10⁻³⁰ cm⁻¹/(molecule cm⁻²) [73] [74] | Varies by molecule and application |
| Key Strengths | Multi-molecular, validated, widely adopted | High accuracy for CO₂, error estimates | Completeness for high-temperature applications |
| Primary Applications | Atmospheric remote sensing, climate studies | CO₂ monitoring, greenhouse gas studies | Exoplanet atmospheres, brown dwarfs, combustion |
Table 2: Methodological Approaches and Data Sources
| Aspect | HITRAN | CDSD | ExoMol |
|---|---|---|---|
| Primary Data Source | Evaluated compilation of experimental & theoretical data | Global fitting to experimental data using effective operators [75] | Ab initio calculations with empirical refinement [76] |
| Position Accuracy | High (validated against laboratory data) | High (experimental positions where possible) [73] | Variable (improved via MARVEL procedure) [76] |
| Uncertainty Quantification | Provided for core parameters | Statistically justified confidence intervals [73] | Explicit uncertainties where available [76] |
| Temperature Treatment | Temperature-dependent parameters | Reference temperature 296 K | Partition functions for extended temperature ranges |
| Updates and Versioning | Quadrennial major releases with interim updates | Periodic updates incorporating new measurements [75] | Continuous updates with new molecules and improvements |
Performance in Specialized Applications
Carbon Dioxide Spectroscopy: For CO₂ specifically, CDSD-296 demonstrates specialized advantages, with most line positions showing differences of only 0.01-0.05 cm⁻¹ compared to the AMES line list [75]. The HITRAN database has incorporated significant portions of CDSD data for its CO₂ line lists, particularly for line positions, while drawing intensities from additional sources including ab initio calculations [77] [75]. This integration strategy leverages the respective strengths of different approaches to optimize overall database performance.
High-Temperature Applications: For high-temperature environments, HITEMP (as part of the HITRAN ecosystem) and ExoMol offer complementary capabilities. The recent HITEMP update for CO₂ implements a novel approach using "effective lines" to account for billions of weak transitions, reducing the number of total lines required for accurate modeling by two orders of magnitude while maintaining accuracy validated up to 2000 K [78]. ExoMol's comprehensive approach makes it particularly valuable for high-temperature applications where complete spectral coverage is more critical than extreme accuracy for individual lines.
Experimental Protocols and Validation Methodologies
Laboratory Validation of Spectroscopic Parameters
The validation of spectroscopic database parameters involves rigorous comparison against high-accuracy laboratory measurements. Cavity Ring-Down Spectroscopy (CRDS) has emerged as a premier technique for these validation studies due to its high sensitivity and accuracy. The typical CRDS experimental protocol involves:
Sample Preparation: Certified reference gases with known composition and isotopologue abundances are introduced into a high-finesse optical cavity [75].
Spectral Acquisition: Laser light is coupled into the cavity, and the ring-down time is measured as a function of wavelength with typical relative uncertainties as low as 0.1% for line intensities [75].
Parameter Retrieval: Line positions, intensities, broadening coefficients, and temperature dependencies are extracted from the measured spectra using multi-spectrum fitting techniques.
Database Comparison: The experimentally determined parameters are compared against database values to identify discrepancies and systematic errors.
These validation protocols have revealed specific areas for improvement across databases. For instance, studies using CRDS and Fourier Transform Spectroscopy (FTS) identified problematic line positions and intensities in HITRAN2016's CO₂ line list in spectral regions between 670 and 8310 cm⁻¹, leading to targeted improvements in subsequent versions [77].
Atmospheric Validation Techniques
Atmospheric measurements provide complementary validation opportunities through real-world observation:
Ground-Based Network Observations: The Total Carbon Column Observing Network (TCCON) and Network for the Detection of Atmospheric Composition Change (NDACC) provide precise atmospheric transmission measurements that serve as validation benchmarks [77].
Solar Occultation Measurements: Instruments like the Atmospheric Chemistry Experiment (ACE) Fourier Transform Spectrometer measure atmospheric transmission using the sun as a source, providing high-resolution reference spectra [77].
Satellite Validation: Data from satellite-based sensors including GOSAT, OCO-2, OCO-3, and MIPAS are compared with synthetic spectra generated using spectroscopic databases, with discrepancies informing database improvements [77].
These atmospheric validation techniques were instrumental in identifying issues with the HITRAN2016 CO₂ line list, particularly in the 1.6 and 2.0 µm regions used for satellite-based CO₂ monitoring, leading to significant improvements in HITRAN2020 [77].
Research Reagent Solutions and Computational Tools
Table 3: Essential Research Tools for Spectroscopic Analysis
| Tool/Resource | Type | Primary Function | Access Method |
|---|---|---|---|
| HAPI (HITRAN Application Programming Interface) | Software Library | Python-based tool for absorption/transmission calculations and data comparison [71] | Download from hitran.org |
| HITRANonline | Web Interface | Primary access point for HITRAN data, cross-sections, and CIA [78] | Web portal (hitran.org) |
| ExoMolHR | Web Interface | Access to high-accuracy empirical lines from ExoMol database [76] | Web portal (exomol.com/exomolhr/) |
| PyExoCross | Software Library | Python program for calculating cross sections from line lists [76] | Download from exomol.com |
| MARVEL Procedure | Algorithm | Measured Active Rotation-Vibration Energy Levels method for empirical energy level determination [76] | Integrated in ExoMol processing |
| Effective Operators Method | Mathematical Framework | Global fitting approach for generating consistent spectroscopic parameters [75] | Core to CDSD methodology |
Future Directions and Development Roadmaps
The field of spectroscopic databases continues to evolve rapidly, with several key trends emerging across the major database initiatives:
HITRAN Development Trajectory: The upcoming HITRAN2024 release demonstrates the project's expansion toward broader planetary and exoplanetary applications, with the addition of "planetary" gases such as S₂, CH₃, and H₃⁺ [72]. The dramatic expansion of cross-section data—adding over 260 molecules not previously represented in HITRAN—significantly extends the database's utility beyond traditional line-by-line applications [72]. Continued development of the HITRAN Application Programming Interface (HAPI) and HAPIEST graphical interface will enhance accessibility for non-specialist users.
ExoMol Advancements: The introduction of ExoMolHR represents a strategic shift toward making the vast ExoMol database more accessible for high-resolution spectroscopic analysis by providing curated, high-accuracy subsets of the full database [76]. The systematic application of the MARVEL procedure to an expanding set of molecules will continue to improve the empirical accuracy of energy levels and transition frequencies. The project's roadmap includes expanding molecular coverage and improving uncertainty quantification across all provided parameters.
CDSD Enhancements: Future developments in CDSD are likely to focus on incorporating new high-accuracy laboratory measurements, particularly from advanced CRDS and frequency comb spectroscopy experiments. Extending the databank to additional temperature regimes and improving the treatment of line-shape parameters represent additional development priorities. The integration of CDSD data with other databases through standardized formats and APIs will enhance interoperability.
These development trajectories collectively point toward increasingly integrated, accessible, and application-specific spectroscopic resources that maintain rigorous standards for accuracy and completeness while serving diverse user communities across atmospheric science, astrophysics, and fundamental molecular physics.
The comparative analysis of HITRAN, CDSD, and ExoMol reveals a sophisticated ecosystem of spectroscopic databases that offer complementary strengths for different scientific applications. HITRAN stands as the most comprehensive multi-molecular database for terrestrial atmospheric applications, with rigorous validation protocols and extensive user support tools. CDSD provides specialized, high-accuracy parameters for carbon dioxide spectroscopy, particularly valuable for greenhouse gas monitoring and climate research. ExoMol offers unparalleled completeness for high-temperature applications, making it essential for exoplanet characterization and astrophysical studies.
The ongoing development of these databases reflects a continuous improvement cycle incorporating new laboratory measurements, advanced theoretical methods, and validation against atmospheric observations. This synergistic relationship between database developers and the broader scientific community ensures that spectroscopic databases continue to evolve in response to emerging scientific challenges and technological capabilities. For researchers engaged in spectroscopic analysis across diverse domains, understanding the distinctive capabilities and appropriate application domains of each database is essential for selecting the optimal resource for specific scientific objectives.
In the field of optoelectronics, the internal architecture of devices such as solar cells and light-emitting diodes is a primary determinant of their performance, stability, and commercial viability. Two principal structures dominate the landscape: the conventional (n-i-p) and the inverted (p-i-n) architectures. The distinction fundamentally lies in the order of the charge-selective layers, which dictates the direction of charge transport and influences everything from fabrication processes to operational degradation mechanisms [79] [80].
This guide provides a technical benchmark of these two architectures, framing the analysis within the context of electron energy level transitions and interfacial spectroscopy. A deep understanding of the energy alignment at the interfaces between the active layer and the charge transport layers is critical, as it governs charge injection, extraction, and recombination dynamics, which ultimately manifest in device-level performance metrics [81] [80].
Architectural Principles and Charge Dynamics
At their core, both conventional and inverted devices consist of an active light-emitting or light-absorbing layer sandwiched between an electron transport layer (ETL) and a hole transport layer (HTL). The sequence of these layers defines the architecture and the resulting charge dynamics.
- Conventional (n-i-p) Architecture: In this structure, the ETL is deposited directly onto the transparent bottom electrode (e.g., ITO), followed by the intrinsic active layer (e.g., perovskite, organic bulk heterojunction), and finally the HTL and the top metal electrode. In solar cells, light typically enters through the ETL side. This architecture requires the ETL to have a conduction band energy that matches both the work function of the cathode and the conduction band of the active layer for efficient electron extraction [82] [80].
- Inverted (p-i-n) Architecture: This configuration reverses the order, placing the HTL on the bottom and the ETL on top. Consequently, the bottom transparent electrode functions as the anode, and the top metal electrode as the cathode. In solar cells, illumination is through the HTL side. For this structure, the energy level alignment requirements are reversed: the HTL's valence band must align with the work function of the anode and the valence band of the active layer [82] [83].
The following diagram illustrates the fundamental charge flow and energy level alignment in these two configurations.
Diagram 1: Charge transport pathways in conventional (n-i-p) and inverted (p-i-n) device architectures. The direction of electron (e⁻) and hole (h⁺) flow is determined by the sequence of the charge transport layers.
The choice of architecture profoundly impacts the recombination zone in light-emitting diodes and the degradation pathways in solar cells. For instance, in single-layer OLEDs with imbalanced charge transport, an inverted structure can shift the recombination zone away from the metallic top electrode, drastically reducing exciton quenching and more than doubling the optical outcoupling efficiency [81].
Performance Benchmarking Across Optoelectronic Devices
The performance of conventional and inverted architectures varies significantly across different material systems and device types. The following tables provide a quantitative benchmark based on recent high-performance demonstrations in organic photovoltaics (OPVs) and perovskite solar cells (PSCs).
Table 1: Performance Benchmark of Organic Photovoltaic (OPV) Architectures
| Architecture | Active Layer System | Highest Reported PCE (%) | Key Stability Metric | Charge Transport Layers |
|---|---|---|---|---|
| Conventional (p-i-n) | PM6:BTP-eC9 (PDSA) [79] | >20 (certified) | N/A | PEDOT:PSS or 2-PACz (HTL); PFN-Br or PDINN (ETL) [79] |
| Inverted (n-i-p) | PM6:BTP-eC9:o-BTP-eC9 (Ternary) [79] | 19.50 (certified 18.97) | >81% PCE after 7,724 h mpp tracking [79] | BHT-capped ZnO (ETL); MoO₃/Ag (HTL/Anode) [79] |
| Inverted (n-i-p) | PM6:Y7-12 (BHJ, slot-die coated) [84] | 15.24 (small area) | High performance retained after 800 h illumination [84] | ZnO (ETL); MoO₃/Ag (HTL/Anode) [84] |
| Inverted (n-i-p) | Small Molecule Donor:Polymer Acceptor (SDPA) [85] | 13.65 | Exceptional thermal stability (84% retention after 72 h at 150°C) [85] | Metal Oxides (e.g., ZnO, MoO₃) [85] |
Table 2: Performance Benchmark of Perovskite Solar Cell (PSC) Architectures
| Architecture | Key Innovation | Highest Reported PCE (%) | Key Stability Metric | Charge Transport Layers |
|---|---|---|---|---|
| Inverted (p-i-n) | CPMAC ionic salt ETL interface [83] | 26.0 | ~95% PCE after 2,200 h at 55°C (mini-module) [83] | CPMAC/C60 (ETL) [83] |
| Conventional (n-i-p) | State-of-the-art with SnO₂ ETL [80] | 25.7 | N/A | SnO₂ (ETL); doped organic HTL (e.g., with LiTFSI) [80] |
| Conventional (n-i-p) | Zr-doped SnO₂ ETL [80] | 19.54 | Improved hysteresis | SnO₂:Zr (ETL) [80] |
| Conventional (n-i-p) | Nb-doped SnO₂ ETL [80] | 20.47 | Low series resistance, better hysteresis | SnO₂:Nb (ETL) [80] |
Analysis of Performance Trends
The benchmark data reveals several key trends:
- Efficiency: In OPVs, conventional architectures currently hold the efficiency record, largely due to advanced polymer-small molecule acceptor systems and effective self-assembled monolayer (SAM) HTLs [79]. In contrast, for PSCs, the highest efficiencies are now comparable between top conventional and inverted cells, with recent inverted devices achieving up to 26% PCE through sophisticated interface engineering [83].
- Stability: Inverted architectures frequently demonstrate superior operational and shelf-life stability. In OPVs, this is attributed to the use of more stable high-work-function metal oxide HTLs (like MoO₃) and air-stable anodes (like Ag), avoiding reactive low-work-function cathodes such as Ca/Al or LiF/Al used in conventional cells [79] [86]. The encapsulation of the metal oxide layers within the device stack also contributes to this robustness [79].
- Manufacturability: Inverted architectures are generally considered more compatible with large-scale, roll-to-roll (R2R) printing and coating methods due to the less thickness-sensitive nature of metal oxide transport layers like ZnO and SnO₂ [79] [84]. Their better environmental robustness simplifies processing requirements for commercial production.
Experimental Protocols for Device Fabrication and Characterization
To ensure reproducible and high-performing devices, standardized protocols for fabrication and characterization are essential. The following sections detail methodologies for key processes.
Protocol: Fabrication of Inverted OPVs with Slot-Die Coating
This protocol, adapted from scalable manufacturing studies, outlines the steps for fabricating inverted OPVs under ambient conditions [84].
- Substrate Preparation: Clean ITO-coated glass substrates sequentially in detergent, deionized water, acetone, and isopropyl alcohol using ultrasonic treatment for 10 minutes each. Dry under a stream of nitrogen gas [86].
- Electron Transport Layer (ETL) Deposition: Deposit a ZnO nanoparticle layer via slot-die coating. The ZnO NPs can be synthesized chemically at room temperature under an oxygen-deficient ambient. Coating parameters (speed, temperature) must be maintained consistently for reproducibility [84] [86].
- Active Layer Deposition: Prepare the photoactive ink, e.g., a blend of polymer donor PM6 and non-fullerene acceptor Y7-12 dissolved in a green solvent like o-xylene. Deposit the active layer using slot-die coating with controlled speed and temperature to form a Bulk Heterojunction (BHJ) film. Alternatively, a Layer-by-Layer (LBL) approach can be employed by sequentially coating the donor and acceptor layers [84] [85].
- Thermal Annealing: Anneal the deposited P3HT:PCBM-based active layer at 150°C for 30 minutes to enhance crystallinity, chain ordering, and π-π stacking, which improves light absorption and charge transport [86].
- Hole Transport Layer (HTL) Deposition: Deposit the HTL, such as MoO₃, via thermal evaporation or a solution-based method.
- Top Electrode Deposition: Finally, thermally evaporate the top metal electrode (e.g., Ag or Al) through a shadow mask to define the active area of the device.
Protocol: Interface Passivation for Inverted PSCs
This protocol describes the application of a novel ionic salt interlayer to strengthen the critical perovskite/ETL interface in inverted PSCs, a key strategy for achieving record efficiencies [83].
- Perovskite Layer Deposition: Deposit the perovskite absorber layer (e.g., via spin-coating or blade-coating) onto the bottom HTL in the inverted p-i-n stack.
- CPMAC Ionic Salt Interlayer Processing:
- Synthesize the CPMAC ionic salt from a reaction of C60 with N-methylglycine, tert-butyl 4-formylbenzylcarbamate, and hydrochloric acid, resulting in 4-(1′,5′-dihydro-1′-methyl-2’H-[5,6]fullereno-C60-Ih-[1,9-c]pyrrol-2′-yl)phenyl-methanaminium chloride [83].
- Deposit a thin layer of CPMAC onto the perovskite surface. This layer acts as an "electron shuttle," fundamentally addressing the weaknesses of a molecular C60 ETL and strengthening the interface by approximately a factor of three [83].
- ETL and Electrode Completion: Complete the device by depositing the C60 ETL and the top metal electrode.
Protocol: Stability and Performance Characterization
Robust characterization is vital for benchmarking.
- Current-Voltage (J-V) Measurements: Perform J-V sweeps under simulated AM 1.5G solar illumination to determine key performance parameters: Power Conversion Efficiency (PCE), Fill Factor (FF), Short-Circuit Current (Jₛ꜀), and Open-Circuit Voltage (Vₒ꜀) [84].
- Maximum Power Point (mpp) Tracking: For stability assessment, operate the device at its mpp under continuous illumination (white light or with specific filters) in controlled environments (e.g., in air or N₂ atmosphere). Monitor the normalized PCE over time. State-of-the-art inverted OPVs have demonstrated T80 lifetimes (time to 80% of initial PCE) exceeding 7,000 hours using this method [79].
- External Quantum Efficiency (EQE): Measure the EQE spectrum to quantify the device's responsivity to different wavelengths of light, which should correlate with the Jₛ꜀ from J-V measurements [84].
- Spatial Performance Mapping: For large-area devices and mini-modules, use Laser Beam Induced Current (LBIC) mapping to visualize spatial variations in photocurrent generation and identify localized defects or recombination losses [84].
The following workflow diagram integrates these fabrication and characterization steps into a coherent experimental pathway.
Diagram 2: Integrated experimental workflow for the fabrication and characterization of conventional and inverted optoelectronic devices, highlighting key steps including interface passivation.
The Scientist's Toolkit: Essential Research Reagents and Materials
The performance of both conventional and inverted architectures is heavily dependent on the materials used for charge transport and interface engineering. The table below catalogs key materials and their functions.
Table 3: Key Research Reagents and Materials for Device Fabrication
| Material Name | Function/Application | Critical Role & Rationale |
|---|---|---|
| ZnO Nanoparticles [79] [84] [86] | Electron Transport Layer (ETL) in inverted OPVs/PSCs. | Provides high electron mobility and solution processability. However, its photocatalytic reactivity under UV light can degrade organic acceptors, necessitating passivation [79]. |
| MoO₃ [79] [86] | Hole Transport Layer (HTL) in inverted architectures; anode interlayer. | A high-work-function, stable metal oxide that replaces acidic and hygroscopic PEDOT:PSS, improving device stability [79] [86]. |
| SnO₂ [79] [80] | Electron Transport Layer (ETL) in conventional and inverted PSCs. | Widely used for its high electron selectivity, optical transparency, and low-temperature processability. Doping (e.g., with Zr, Nb) enhances its conductivity and reduces hysteresis [80]. |
| PEDOT:PSS [79] [80] | Hole Transport Layer (HTL) in conventional architectures. | A common conductive polymer HTL. Its acidic and hygroscopic nature can corrode ITO and degrade device stability over time, a key weakness of conventional structures [79] [80]. |
| BHT (3-(3,5-di-tert-butyl-4-hydroxyphenyl)propionic acid) [79] | Passivation agent for ZnO ETL. | Forms COO⁻Zn complexes, passivating surface oxygen vacancies. Acts as a radical scavenger to suppress light-induced decomposition of non-fullerene acceptors, boosting both efficiency and stability [79]. |
| CPMAC Ionic Salt [83] | Interface modifier for ETL in inverted PSCs. | A C60-based ionic salt that strengthens the perovskite/ETL interface, suppresses ion migration, and significantly enhances thermal and operational stability while enabling high efficiency [83]. |
| PFN-Br / PDINN [79] | Electron Transport Layer (ETL) in conventional OPVs. | Polymer-based interfacial materials that provide favorable work function modification and improve electron injection/extraction in conventional structures [79]. |
| 2-PACz [79] | Hole Transport Layer (HTL) in conventional OPVs. | A self-assembled monolayer (SAM) HTL that enables high-efficiency conventional OPVs by providing excellent hole selectivity and energy level alignment [79]. |
The benchmark analysis clearly shows that the competition between conventional and inverted architectures is context-dependent, dictated by the specific material system and target application. Conventional n-i-p architectures in OPVs currently lead in pure efficiency metrics, benefiting from highly optimized organic charge transport layers and active materials. Conversely, inverted p-i-n architectures consistently demonstrate superior operational stability, better compatibility with large-scale manufacturing, and, in the case of PSCs, have recently matched or even surpassed the efficiency of conventional cells through innovative interface engineering.
The central theme uniting advancements in both architectures is the critical importance of interface management. Defects, photocatalytic reactivity, and energetic misalignment at the interfaces between the active layer and charge transport layers are the primary bottlenecks for both performance and stability. Spectroscopic techniques that probe electron energy level transitions are therefore indispensable for diagnosing interfacial losses and guiding material design. Future progress hinges on the continued development of multifunctional interfacial materials and passivation strategies that can simultaneously optimize electronic structure, suppress chemical degradation, and enhance mechanical adhesion, thereby pushing both conventional and inverted devices toward their theoretical limits and eventual commercial realization.
The selection between Single-Photon Emission Computed Tomography (SPECT) and Positron Emission Tomography (PET) represents a critical decision point in nuclear medicine, fundamentally rooted in the distinct electron energy level transitions of their respective radionuclides. Both modalities enable the non-invasive visualization of biological processes by detecting radiation emitted from radiopharmaceuticals administered to patients [87]. The core difference stems from the underlying nuclear decay processes: SPECT utilizes radionuclides that decay via single gamma-ray emission, whereas PET relies on radionuclides that decay via positron emission, leading to annihilation and the production of two coincident 511-keV photons [48] [88].
This physical distinction directly parallels concepts in electron energy level transitions spectroscopy. In SPECT, the single gamma-ray emission is analogous to a single electronic transition event. In PET, the positron annihilation process represents a more complex event where the positron (antimatter electron) interacts with an electron, resulting in their mass-energy conversion into two photons [88]. The energy of these photons (511 keV each) is a direct consequence of the electron-positron mass-energy equivalence according to E=mc², a fundamental physical constant. Understanding these decay pathways and their associated energies is essential for selecting the appropriate radionuclide and imaging modality for specific diagnostic applications.
Physical Principles and Decay Mechanisms
SPECT: Single Gamma-Ray Emission
SPECT imaging is based on the detection of single gamma-ray photons emitted from radionuclides. The imaging chain begins with a radiopharmaceutical administered to the patient that contains a gamma-emitting isotope. As the isotope decays, it emits single photons of characteristic energy [87]. A gamma camera detects these photons, utilizing key components including a collimator to define the direction of incoming photons, a sodium iodide scintillation crystal to convert gamma energy to light, and an array of photomultiplier tubes to amplify and locate the light events [89] [87].
The collimator is a critical determinant of SPECT image quality and sensitivity, with different geometries (parallel-hole, converging, diverging, pinhole) available for specific clinical applications [87]. However, the collimator necessarily absorbs the vast majority of emitted photons, resulting in low detection efficiency—a fundamental limitation of SPECT technology [89].
PET: Positron Emission and Annihilation
PET imaging exploits a more complex physical process beginning with positron emission from proton-rich radionuclides. The emitted positron travels a short distance in tissue (the positron range, which depends on its energy), losing kinetic energy until it annihilates with an electron [88]. This annihilation event converts the combined mass of the positron and electron into two 511-keV gamma photons emitted in nearly opposite directions (180 degrees apart) [48] [88].
PET scanners detect these photon pairs through coincidence timing. When two detectors register photons within a very narrow time window (typically a few nanoseconds), the annihilation event is localized to a line connecting the two detectors [89]. This coincidence detection method provides inherent spatial localization and eliminates the need for physical collimation, contributing to PET's significantly higher sensitivity compared to SPECT [89]. Modern PET systems further improve image resolution through "Time of Flight" (TOF) technology, which uses the tiny difference in arrival times of the two photons to better localize the annihilation event along the line of response [89].
Diagram 1: Fundamental imaging processes for PET and SPECT technologies.
Radionuclide Characteristics and Applications
Common Radionuclides in Clinical Use
The selection of radionuclides for SPECT and PET imaging is determined by their physical properties, availability, and compatibility with targeting molecules. Table 1 summarizes the key radionuclides used in clinical practice.
Table 1: Common Radionuclides for SPECT and PET Imaging
| Modality | Radionuclide | Half-Life | Primary Energy | Production Method | Key Clinical Applications |
|---|---|---|---|---|---|
| SPECT | Technetium-99m (⁹⁹mTc) | 6.03 hours | 140 keV | Generator | Bone scans, myocardial perfusion, renal imaging |
| SPECT | Iodine-123 (¹²³I) | 13.2 hours | 159 keV | Cyclotron | Thyroid imaging, neuroendocrine tumors |
| SPECT | Indium-111 (¹¹¹In) | 2.8 days | 171, 245 keV | Cyclotron | Infection imaging, somatostatin receptor scintigraphy |
| PET | Fluorine-18 (¹⁸F) | 109.8 minutes | 511 keV (annihilation) | Cyclotron | Oncology ([¹⁸F]FDG), neuroimaging ([¹⁸F] amyloid tracers) |
| PET | Gallium-68 (⁶⁸Ga) | 68 minutes | 511 keV (annihilation) | Generator | Neuroendocrine tumors ([⁶⁸Ga]Ga-DOTA-TATE), prostate cancer ([⁶⁸Ga]Ga-PSMA-11) |
| PET | Carbon-11 (¹¹C) | 20.4 minutes | 511 keV (annihilation) | Cyclotron | Neurotransmitter studies, receptor imaging |
| PET | Zirconium-89 (⁸⁹Zr) | 78.4 hours | 511 keV (annihilation) | Cyclotron | Immuno-PET with monoclonal antibodies |
Practical Considerations for Radionuclide Selection
Several practical factors influence radionuclide selection for diagnostic applications:
Half-life matching: The radionuclide's physical half-life must be compatible with the biological half-life of the targeting process. For example, ⁸⁹Zr (half-life 78.4 hours) is ideally suited for immuno-PET with monoclonal antibodies, which require days to achieve optimal target-to-background ratios [90]. Conversely, ¹¹C (half-life 20.4 minutes) is restricted to imaging targets with very rapid uptake, such as neurotransmitter receptors [88].
Production and availability: SPECT radionuclides like ⁹⁹mTc are available from generators, enabling use in facilities without cyclotrons [91]. PET radionuclides typically require cyclotron production, though generator-produced isotopes like ⁶⁸Ga have increased PET accessibility [48].
Image quality requirements: PET generally provides superior image resolution and sensitivity due to its higher photon flux and absence of physical collimation [89] [91]. SPECT remains valuable for applications where its lower resolution is sufficient, or when specific SPECT radionuclides offer unique biological targeting.
Quantification capabilities: PET offers more robust quantitative capabilities, with standardized uptake values (SUV) widely used for treatment response assessment in oncology [92]. While quantitative SPECT is possible, it is more challenging to implement and less standardized [93].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 2: Key Research Reagents and Materials for SPECT/PET Radiochemistry
| Item | Function | Application Examples |
|---|---|---|
| Chelators | Bind radiometals to targeting vectors | DOTA, NOTA, DFO for ⁶⁸Ga, ¹⁷⁷Lu, ⁸⁹Zr labeling [90] [48] |
| Precursors | Chemical substrates for radiolabeling | Precursor molecules for nucleophilic substitution (¹⁸F) or electrophilic substitution [88] |
| Targeting Vectors | Provide specificity for biological targets | Peptides, antibodies, small molecules, engineered scaffold proteins [90] [48] |
| Quality Control Systems | Ensure radiopharmaceutical safety and purity | HPLC, TLC, GC systems for chemical and radiochemical purity testing [88] |
| Dose Calibrators | Measure radioactivity concentration | Essential for quantitative imaging and patient dose administration [93] |
| Imaging Phantoms | Validate scanner performance | NEMA NU-2 IEC Image Quality phantom for quantification accuracy [93] |
Experimental Protocols and Validation Methodologies
Quantitative Imaging Validation Protocol
Multicenter clinical trials requiring quantitative imaging necessitate rigorous validation of both PET and SPECT systems. The Australasian Radiopharmaceutical Trials Network (ARTnet) has established a robust three-step methodology for this purpose [93]:
Initial Calibration Verification: Confirm quantitative accuracy using a standard radionuclide (e.g., ¹⁸F for PET, ⁹⁹mTc for SPECT) in a simple geometric phantom (e.g., 20-cm diameter cylinder).
Novel Radionuclide Testing: Validate quantitative accuracy using the trial-specific radionuclide (e.g., ¹²⁴I for PET, ¹³¹I for SPECT) in the same simple phantom.
Complex Phantom Validation: Assess performance using the novel radionuclide in an anthropomorphic phantom (NEMA NU-2 IEC Image Quality phantom) to derive metrics including quantitative accuracy based on standardized uptake values (SUV) [93].
This protocol ensures that reconstructed radioactivity concentration (Bq/cm³) measurements are accurate across different imaging systems and sites, enabling reliable data pooling in multicenter trials.
Diagram 2: Three-step quantitative imaging validation protocol for clinical trials.
Radiopharmaceutical Development Workflow
The development of novel radiopharmaceuticals follows a structured pathway from concept to clinical application:
Target Identification: Selection of a biologically relevant molecular target with appropriate expression patterns in the disease state.
Vector Selection: Choice of targeting moiety (small molecule, peptide, antibody, engineered protein) based on pharmacokinetic requirements and target biology [90] [48].
Radionuclide Matching: Pairing of radionuclide with appropriate decay properties to the biological half-life of the vector-target interaction.
Radiolabeling Optimization: Development of efficient radiochemistry for incorporating the radionuclide into the targeting vector while maintaining biological activity [88].
Preclinical Validation: In vitro and in vivo assessment of binding affinity, specificity, pharmacokinetics, and dosimetry.
Clinical Translation: Implementation under Good Manufacturing Practice (GMP) conditions for human studies [88].
Emerging Trends and Future Perspectives
Theranostics: Integrating Diagnosis and Therapy
The concept of "theranostics" represents a paradigm shift in nuclear medicine, pairing diagnostic and therapeutic radionuclides targeting the same biomarker [92] [48]. Notable examples include:
- Neuroendocrine Tumors: [⁶⁸Ga]Ga-DOTA-TATE for PET imaging paired with [¹⁷⁷Lu]Lu-DOTA-TATE for therapy [48].
- Prostate Cancer: [⁶⁸Ga]Ga-PSMA-11 for PET imaging paired with [¹⁷⁷Lu]Lu-PSMA-617 for therapy [48].
This approach enables patient-specific treatment planning, where diagnostic imaging identifies candidates for radionuclide therapy and predicts treatment response.
Technological Advancements
Both SPECT and PET technologies continue to evolve through several key developments:
Hardware Improvements: Digital PET detectors, cadmium zinc telluride (CZT) semiconductor detectors for SPECT, and total-body PET systems that dramatically increase sensitivity [92] [94].
Radiochemistry Innovations: Novel labeling methods for difficult substrates, development of bifunctional chelators with improved stability, and labeling strategies for new radionuclides [90] [88].
Quantitative Analysis Tools: Artificial intelligence-based image reconstruction and analysis, standardized reporting protocols, and advanced kinetic modeling techniques [92] [91].
Table 3: Comparison of Key Characteristics Between SPECT and PET Imaging
| Parameter | SPECT | PET |
|---|---|---|
| Physical Principle | Single gamma-ray detection | Coincidence detection of annihilation photons |
| Sensitivity | Low (collimator-limited) | High (collimator-free) |
| Spatial Resolution | 8-12 mm (clinical) | 4-6 mm (clinical) |
| Quantitative Accuracy | Moderate | High |
| Common Radionuclides | ⁹⁹mTc, ¹²³I, ¹¹¹In | ¹⁸F, ⁶⁸Ga, ¹¹C, ⁸⁹Zr |
| Radionuclide Availability | Generator (⁹⁹mTc), cyclotron | Primarily cyclotron, some generators (⁶⁸Ga) |
| Typical Scan Time | 15-30 minutes | 10-20 minutes |
| Equipment Cost | Lower | Higher |
| Primary Clinical Domains | Cardiology, bone imaging, perfusion studies | Oncology, neurology, cardiology |
The selection between SPECT and PET imaging technologies represents a critical decision in diagnostic nuclear medicine, with implications for image quality, quantitative accuracy, clinical workflow, and patient management. This choice is fundamentally guided by the physical decay properties of available radionuclides and their compatibility with biological targeting vectors. While PET offers superior sensitivity and quantification capabilities, SPECT maintains important roles in clinical practice due to its wider availability, lower cost, and established clinical applications in cardiology and general nuclear medicine.
The ongoing evolution of both modalities—driven by innovations in hardware, radiochemistry, and the growing importance of theranostics—ensures that both SPECT and PET will continue to play complementary roles in molecular imaging. For researchers and clinicians, the optimal selection between these modalities requires careful consideration of the specific clinical or research question, available infrastructure, and the pharmacokinetics of the biological process under investigation.
In the field of electron energy level transitions spectroscopy, cross-platform validation represents a critical methodology for ensuring the reliability and accuracy of research findings. This process involves the systematic integration of theoretical calculations with experimental data to create a robust, self-consistent scientific framework. For researchers investigating fundamental atomic properties, electron affinities, and electronic structures, this validation paradigm is particularly crucial when studying rare, radioactive, or superheavy elements where experimental data is exceptionally limited [95]. The convergence of advanced spectroscopic techniques with sophisticated computational models enables scientists to push the boundaries of atomic physics and materials science, providing unprecedented insights into electron correlation effects and electronic behaviors in functional materials [96].
Fundamental Principles of Cross-Platform Validation
Core Conceptual Framework
Cross-platform validation in spectroscopy research operates on the principle of methodological triangulation, where multiple independent approaches are employed to investigate the same fundamental phenomena. The convergence of results from different methodologies significantly strengthens scientific conclusions. This approach is particularly valuable in domains where systematic errors may affect individual techniques [95]. For electron energy level research, this typically involves correlating data from photoemission spectroscopy, laser photodetachment experiments, and theoretical many-body calculations.
The validation framework relies on establishing quantitative metrics for comparing results across different platforms. These include energy level measurements, cross-section determinations, spectral line shapes, and intensity ratios. Statistical measures of agreement, such as confidence intervals and standard deviations, provide objective criteria for validation success [97]. When discrepancies arise, they often point to underlying physical phenomena not accounted for in existing models, potentially driving theoretical advancements.
Challenges in Spectroscopic Validation
Several significant challenges complicate cross-platform validation in electron spectroscopy. Technical variations in experimental setups, including differing energy resolutions, photon fluxes, and detection efficiencies, can create apparent inconsistencies between datasets. Similarly, computational approximations in theoretical methods, such as basis set limitations, correlation treatment, and relativistic effects, introduce uncertainties that must be quantified [95] [96].
For rare elements and radioactive specimens, sample limitations present additional hurdles. Production rates for some exotic species can be as low as a few atoms per second, severely constraining experimental options and statistical precision [95]. Furthermore, material quality profoundly impacts results, as defects, impurities, and structural imperfections can significantly alter electronic properties [96].
Experimental Methodologies for Electron Energy Level Analysis
Laser Photodetachment Threshold Spectroscopy
Laser Photodetachment Threshold (LPT) spectroscopy serves as a powerful technique for determining electron affinities with high precision. The fundamental principle involves exposing negatively-charged ions to laser photons, resulting in detachment of the extra electron when photon energy exceeds the electron binding energy. The experimental signature is a sharp increase in neutral atom production as the laser energy crosses the detachment threshold [95].
Recent methodological innovations have dramatically enhanced LPT sensitivity. The MIRACLS approach (Multi-Ion Reflection Apparatus for Collinear Laser Spectroscopy) confines ions in an electrostatic trap between electrostatic mirrors, significantly increasing laser interaction time. This enables measurements with approximately five orders of magnitude fewer anions than conventional techniques while maintaining state-of-the-art precision. The method's collinear geometry compresses the velocity spread of anions, reducing Doppler broadening and improving spectral resolution [95].
The critical experimental parameters for LPT spectroscopy include:
- Laser specifications: Narrow-bandwidth continuous-wave lasers reduce uncertainties from broadening effects
- Ion beam properties: Low emittance bunches with well-defined kinetic energies
- Detection system: High-efficiency, low-background neutral particle detectors
- Environmental control: Ultra-high vacuum conditions to minimize unwanted interactions
Synchrotron Radiation Photoemission Spectroscopy
Synchrotron radiation photoemission spectroscopy provides element-specific electronic state information with exceptional resolution. This technique utilizes tunable, high-brightness X-rays from synchrotron facilities to eject electrons from specific core levels, enabling detailed analysis of electronic density of states [96].
The methodology's unique strength lies in tuning X-ray energy to match absorption edges of specific elements, thereby enhancing signal from targeted electron orbitals. For functional oxide materials like SrRuO₃, this enables separate examination of partial density of states derived from cation (e.g., Ru 4d) and anion (e.g., O 2p) orbitals. Recent experiments using this approach have revealed unexpectedly strong electron correlations in oxygen orbitals, challenging conventional theoretical models that primarily considered electron interactions in transition metals [96].
Key experimental considerations include:
- Sample quality: Atomically ordered thin films fabricated using advanced techniques like machine learning-optimized molecular beam epitaxy
- Energy resolution: Monochromatized X-ray sources with precise energy calibration
- Detection geometry: Angle-resolved setups for band structure determination
- Temperature control: Cryogenic systems for studying temperature-dependent effects
Comparative Analysis of Spectroscopic Techniques
Table 1: Comparison of Spectroscopic Methods for Electron Energy Level Analysis
| Method | Physical Principle | Energy Resolution | Element Specificity | Sample Requirements | Primary Applications |
|---|---|---|---|---|---|
| Laser Photodetachment Threshold Spectroscopy | Photon-induced electron detachment | Very High (meV range) | Moderate | Anion beams, ~10⁶ particles/second | Electron affinity measurements, atomic anion properties |
| Synchrotron Radiation Photoemission | Photoelectric effect | High (10-100 meV) | Excellent (tunable) | High-quality thin films, single crystals | Partial density of states, electron correlations, band structure |
| Photodetachment Microscopy | Quantum interference patterns | Ultra-High (μeV range) | Moderate | Collimated anion beams | Precise electron affinities, quantum interference |
| Velocity Map Imaging | Electron kinetic energy mapping | High | Moderate | Gas-phase ions | Photoelectron angular distributions, kinetic energy spectra |
Theoretical Frameworks and Computational Approaches
Many-Body Quantum Methods
Theoretical interpretation of electron energy level transitions relies heavily on many-body quantum methods that account for complex electron-electron correlations. These approaches move beyond simple independent-electron models to describe how electrons collectively behave in atoms and materials [95]. For heavy elements and functional materials, fully relativistic implementations are essential to accurately capture spin-orbit coupling and other relativistic effects that significantly influence electronic structure [95].
Advanced computational frameworks include:
- Configuration interaction methods: Accounting for electron correlation through linear combinations of Slater determinants
- Coupled cluster theory: Systematic inclusion of electron excitations for high-precision atomic calculations
- Dynamical mean-field theory (DMFT): Treating local electron correlations in materials, particularly valuable for strongly correlated systems
- Density functional theory (DFT) + U: Incorporating strong on-site correlations in transition metal oxides
These theoretical models require validation against precisely measured spectroscopic data, creating a feedback loop where experimental results refine computational approaches, which in turn predict new phenomena to test experimentally.
Benchmarking and Uncertainty Quantification
Rigorous benchmarking procedures establish the reliability of theoretical methods by comparing predictions with high-precision experimental measurements. Well-characterized systems with extensive experimental data, such as chlorine atoms, provide critical reference points for validating computational approaches before applying them to less-studied elements [95].
Uncertainty quantification represents an essential component of theoretical frameworks. This includes:
- Numerical uncertainties: From basis set incompleteness, grid integrations, and convergence thresholds
- Methodological uncertainties: From approximations in electron correlation treatment and relativistic formalisms
- Physical uncertainties: From neglected effects such as quantum electrodynamics contributions
Effective cross-platform validation requires transparent reporting of all significant uncertainty components from both experimental and theoretical approaches, enabling meaningful comparison and reconciliation of results.
Integrated Workflow for Cross-Platform Validation
Figure 1: Integrated validation workflow showing the iterative process of combining theoretical and experimental approaches.
Essential Research Reagent Solutions
Table 2: Key Research Reagents and Materials for Electron Spectroscopy
| Reagent/Material | Function | Specification Requirements | Application Examples |
|---|---|---|---|
| Strontium Ruthenate (SrRuO₃) Thin Films | Model functional oxide material | Atomically ordered perovskite structure, machine learning-optimized growth | Electronic structure studies, electron correlation measurements [96] |
| Chlorine Anion Beams | Benchmark species for method validation | High purity (³⁵Cl⁻ selection), low emittance, controlled kinetic energy | Electron affinity measurements, technique calibration [95] |
| Synchrotron Radiation | High-brightness tunable X-ray source | Energy tunability (50-2000 eV), high flux, polarization control | Element-specific density of states measurements [96] |
| Narrow-Bandwidth Lasers | Precision photodetachment source | Continuous-wave operation, narrow linewidth (<1 MHz), wavelength stability | Threshold spectroscopy, reduced Doppler broadening [95] |
| Ultra-High Vacuum Systems | Experimental environment control | Base pressure <10⁻¹⁰ mbar, minimal hydrocarbon contamination | Surface-sensitive measurements, impurity reduction |
| Electrostatic Ion Traps | Anion storage and manipulation | High mirror potentials, stable field configurations, precise timing control | Multi-reflection experiments, extended laser interaction [95] |
Data Presentation and Analysis Framework
Standards for Quantitative Data Presentation
Effective data presentation is fundamental to cross-platform validation, enabling clear comparison between theoretical and experimental results. Tables should be self-explanatory, with descriptive titles, clearly labeled units, and appropriate organization that facilitates comparison of related values [98] [97].
For spectroscopic data, recommended practices include:
- Normalization procedures: Consistent scaling of intensities to enable meaningful comparisons
- Uncertainty reporting: Explicit documentation of statistical and systematic errors for all measured values
- Reference standards: Inclusion of well-characterized benchmark systems in data tables
- Data transformation documentation: Clear description of any processing applied to raw data
Table 3: Electron Affinity Measurements and Theoretical Predictions for Benchmark Elements
| Element | Experimental EA (eV) | Uncertainty (±eV) | Theoretical EA (eV) | Method | Deviation (%) |
|---|---|---|---|---|---|
| Chlorine (³⁵Cl) | 3.612720 | 0.000044 | 3.613 | CCSD(T) | 0.008 |
| Astatine | 2.4158 | 0.0007 | 2.465 | Relativistic CI | 2.0 |
| Oxygen | 1.461 | 0.006 | 1.454 | MR-CI | 0.5 |
| Strontium Ruthenate | - | - | - | DFT+DMFT | - |
Visualization Principles for Spectral Data
Effective data visualization enables immediate recognition of key spectral features and validation outcomes. Adherence to fundamental design principles ensures clarity and interpretability [98]:
- Color contrast: Sufficient luminance difference between plot elements and backgrounds (minimum 4.5:1 for standard text, 3:1 for large text) [99] [100]
- Axis labeling: Complete descriptions with units and measurement conditions
- Legend design: Clear identification of different data series and theoretical curves
- Uncertainty visualization: Error bars or confidence intervals representing measurement precision
For composite figures showing both experimental and theoretical results, consistent color coding helps distinguish data sources (e.g., blue for experimental results, red for theoretical calculations), with explicit documentation in figure captions.
Case Study: Oxygen Electron Correlations in Strontium Ruthenate
Figure 2: Research pathway leading to the discovery of orbital-dependent electronic states in SrRuO₃.
A recent breakthrough in functional oxides research exemplifies the power of cross-platform validation. Investigation of strontium ruthenate (SrRuO₃) combining synchrotron radiation photoemission spectroscopy with theoretical analysis revealed unexpected orbital-dependent electron correlations [96].
The experimental approach utilized:
- Machine learning-optimized molecular beam epitaxy to fabricate ultrahigh-quality thin films with atomic-level ordering
- Tunable synchrotron radiation to selectively probe partial density of states from ruthenium 4d and oxygen 2p orbitals
- Advanced spectral analysis to quantify electron correlation strengths through comparison of Auger spectra and self-convolution calculations
Results demonstrated that while Ru 4d orbitals exhibit metallic character with significant density of states at the Fermi energy, O 2p orbitals show insulating behavior with nearly zero density of states at EF. This unexpected divergence from conventional models, which assumed strongly hybridized orbitals with similar shapes, was attributed to strong electron correlation in oxygen orbitals several times larger than in ruthenium orbitals [96].
This case study highlights how sophisticated experimental techniques, coupled with critical assessment against theoretical predictions, can overturn established paradigms and reveal previously overlooked physical mechanisms determining material properties.
The continuing evolution of cross-platform validation methodologies promises to address increasingly complex challenges in electron energy level research. Several emerging trends are particularly noteworthy:
Sensitivity-enhanced techniques like the MIRACLS approach will enable exploration of increasingly rare systems, potentially extending to superheavy elements where relativistic effects dominate electronic behavior [95]. The ability to perform precise measurements with minute quantities of material opens unprecedented opportunities for studying elements previously inaccessible to experimental characterization.
Machine learning integration across both experimental and theoretical domains represents another transformative development. ML-optimized material synthesis [96], automated data analysis, and enhanced computational methods will accelerate the validation cycle and improve predictive capabilities.
The ongoing development of theoretical frameworks that more comprehensively treat electron correlations, particularly in complex materials and heavy elements, will provide sharper tools for interpreting experimental results. Conversely, increasingly precise experimental data will create more rigorous benchmarks for theoretical advancements.
In conclusion, cross-platform validation integrating theoretical calculations with experimental data establishes a robust foundation for advancing our understanding of electron energy level transitions. This multidisciplinary approach, leveraging complementary strengths of various spectroscopic and computational techniques, enables researchers to transcend the limitations of individual methods and achieve unprecedented insights into electronic structure and behavior.
Conclusion
Mastering electron energy level transitions is fundamental to advancing spectroscopic applications in biomedical and clinical research. The journey from foundational quantum principles to sophisticated applications in theranostics and pharmaceutical analysis demonstrates the transformative power of this knowledge. Future directions point toward the increased integration of machine learning and artificial intelligence to handle complex spectral datasets, the development of novel radiopharmaceuticals for targeted alpha therapy, and the design of next-generation, flexible organic photodetectors for portable diagnostic tools. By continuing to refine detection methods for weak transitions and validate data with increasing precision, spectroscopy will remain a cornerstone technology for enabling personalized medicine, improving drug discovery pipelines, and unlocking new frontiers in non-invasive disease diagnosis and treatment.
References
- 1. Ground State Excited States - Electronic Structure - MCAT ... [https://jackwestin.com/resources/mcat-content/electronic-structure/ground-state-excited-states]
- 2. Emission Spectra and Energy (1.3.1) | IB DP... | TutorChase Levels [https://www.tutorchase.com/notes/ib/chemistry-2025-hl/1-3-1-emission-spectra-and-energy-levels]
- 3. 29.1 Quantization of Energy – College Physics: OpenStax [https://pressbooks.bccampus.ca/collegephysics/chapter/quantization-of-energy/]
- 4. Spectroscopy Is Applied Quantum Mechanics, Part II [https://www.spectroscopyonline.com/view/spectroscopy-applied-quantum-mechanics-part-ii-quantum-revolution]
- 5. Understanding Quantized Energy Levels in Atoms [https://www.physicsclassroom.com/Chemistry-Tutorial/Modern-Atomic-Model/Bohrs-Quantized-Energy-Levels]
- 6. Theory of electronic | transitions Class Notes | Fiveable Spectroscopy [https://fiveable.me/spectroscopy/unit-4/theory-electronic-transitions/study-guide/9SgNHJVlKvKzsC4O]
- 7. Atomic Spectra: Key Concepts, Formulas & Applications [https://www.vedantu.com/physics/atomic-spectra]
- 8. Basic Concepts in Fluorescence [https://evidentscientific.com/en/microscope-resource/knowledge-hub/techniques/fluorescence/fluorescenceintro]
- 9. Fluorescence Fundamentals | Thermo Fisher Scientific - US [https://www.thermofisher.com/us/en/home/references/molecular-probes-the-handbook/introduction-to-fluorescence-techniques.html]
- 10. Fluorescence Emission, Absorption, & Excitation | Angstrom [https://angtech.com/understanding-absorption-emission-and-excitation-in-fluorescence/]
- 11. Basic Principles Of Fluorescence And Ion Sensing [https://www.ionoptix.com/resource/basic-principles-of-fluorescence-and-ion-sensing/]
- 12. An excitation-wavelength-dependent organic ... [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d4sc08197h]
- 13. Fluorescence excitation strategies driven by different ... [https://www.sciencedirect.com/science/article/abs/pii/S100184172500734X]
- 14. Selection rule [https://en.wikipedia.org/wiki/Selection_rule]
- 15. Selection Rule - an overview | ScienceDirect Topics [https://www.sciencedirect.com/topics/mathematics/selection-rule]
- 16. Laporte rule [https://en.wikipedia.org/wiki/Laporte_rule]
- 17. 11.3.1: Selection Rules [https://chem.libretexts.org/Bookshelves/Inorganic_Chemistry/Inorganic_Chemistry_(LibreTexts)/11%3A_Coordination_Chemistry_III_-_Electronic_Spectra/11.03%3A_Electronic_Spectra_of_Coordination_Compounds/11.3.01%3A_Selection_Rules]
- 18. La Porte Selection rule.pptx [https://www.slideshare.net/slideshow/la-porte-selection-rulepptx/253067144]
- 19. Laporte Selection Rule - (Inorganic Chemistry I) [https://fiveable.me/key-terms/inorganic-chemistry-i/laporte-selection-rule]
- 20. 18.4 Electronic spectroscopy - Physical Chemistry I [https://fiveable.me/physical-chemistry-i/unit-18/electronic-spectroscopy/study-guide/NRMj1SHxUdWwq3ee]
- 21. Recent advances in rare earth oxides applied to energy ... [https://www.sciencedirect.com/science/article/abs/pii/S0272884224056554]
- 22. Electronic Spectroscopy - Interpretation [https://chem.libretexts.org/Bookshelves/Physical_and_Theoretical_Chemistry_Textbook_Maps/Supplemental_Modules_(Physical_and_Theoretical_Chemistry)/Spectroscopy/Electronic_Spectroscopy/Electronic_Spectroscopy_-_Interpretation]
- 23. Rotational–vibrational spectroscopy [https://en.wikipedia.org/wiki/Rotational%E2%80%93vibrational_spectroscopy]
- 24. Rovibrational Spectroscopy [https://chem.libretexts.org/Bookshelves/Physical_and_Theoretical_Chemistry_Textbook_Maps/Supplemental_Modules_(Physical_and_Theoretical_Chemistry)/Spectroscopy/Rotational_Spectroscopy/Rovibrational_Spectroscopy]
- 25. Rotational–vibrational coupling [https://en.wikipedia.org/wiki/Rotational%E2%80%93vibrational_coupling]
- 26. Precision Spectroscopy of the Fine Structure in the 𝒂 𝚺 𝒖 ... [https://arxiv.org/html/2508.08060v2]
- 27. Quantitative Infrared Database - the NIST WebBook [https://webbook.nist.gov/chemistry/quant-ir/]
- 28. A Survey of Chemometric Methods Used in Spectroscopy [https://www.spectroscopyonline.com/view/a-survey-of-chemometric-methods-used-in-spectroscopy]
- 29. Web Sources - Spectra and Spectral Data [https://guides.lib.utexas.edu/chemistry/spectra]
- 30. Electron Transitions in Optical Emission and X-Ray Fluorescence Spectrometry | Spectroscopy Online [https://www.spectroscopyonline.com/view/electron-transitions-optical-emission-and-x-ray-fluorescence-spectrometry]
- 31. 8.6: Atomic Spectra and X-rays [https://phys.libretexts.org/Bookshelves/University_Physics/University_Physics_(OpenStax)/University_Physics_III_-_Optics_and_Modern_Physics_(OpenStax)/08%3A_Atomic_Structure/8.06%3A_Atomic_Spectra_and_X-rays]
- 32. Optical and x-ray technology synergies enabling diagnostic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6197862/]
- 33. X-Ray Absorption and Emission Spectroscopy in ... [https://www.mdpi.com/2076-3417/15/19/10784]
- 34. X-ray Emission From Atoms - Imagine the Universe! - NASA [https://imagine.gsfc.nasa.gov/science/toolbox/xray_generation_atom.html]
- 35. High-resolution X-ray spectroscopy of rare events [https://pmc.ncbi.nlm.nih.gov/articles/PMC4028048/]
- 36. Spectroscopy of L-series transitions for x-ray laser ... [https://opg.optica.org/abstract.cfm?uri=josab-2-12-1894]
- 37. Characterization and DNA binding studies of unexplored imidazolidines by electronic absorption spectroscopy and cyclic voltammetry - PubMed [https://pubmed.ncbi.nlm.nih.gov/23474470/]
- 38. Inelastic Electron Tunneling Spectroscopy of Molecular Electronic Junctions: Recent Advances and Applications [https://www.mdpi.com/2073-4352/15/8/681]
- 39. Electron Spectroscopy and Related Methods [https://link.springer.com/chapter/10.1007/978-1-4684-4997-6_7]
- 40. Electronic Absorption Spectroscopy - an overview [https://www.sciencedirect.com/topics/chemistry/electronic-absorption-spectroscopy]
- 41. Application and Method of Surface Plasmon Resonance ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11249113/]
- 42. Near-infrared spectroscopy and imaging: Basic principles ... [https://www.sciencedirect.com/science/article/abs/pii/S0169409X05000578]
- 43. Near-infrared spectroscopy [https://en.wikipedia.org/wiki/Near-infrared_spectroscopy]
- 44. Electronic theory of Near Infra-Red (NIR) chemistry? [https://www.ssi.shimadzu.com/service-support/faq/uv-vis/light-and-theory/19/index.html]
- 45. NIR vs IR: What is the difference? [https://www.metrohm.com/en/discover/blog/2024/nir-vs-ir.html]
- 46. Near-infrared Spectroscopy Market Size & Share Analysis [https://www.mordorintelligence.com/industry-reports/near-infrared-spectroscopy-market]
- 47. Recent applications of near infrared to pharmaceutical ... [https://pubmed.ncbi.nlm.nih.gov/40413709/]
- 48. Radiopharmaceuticals and their applications in medicine [https://www.nature.com/articles/s41392-024-02041-6]
- 49. Activity quantification and dosimetry in radiopharmaceutical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11440950/]
- 50. Noise Suppression in Organic Photodiodes [https://pubmed.ncbi.nlm.nih.gov/41268801/]
- 51. Peculiarities of room temperature organic photodetectors [https://www.nature.com/articles/s41377-025-01939-2]
- 52. Electron, hole, and energy transfer dynamics in non-fullerene ... [https://pubs.rsc.org/doi/d4sc04072d]
- 53. Doping effects for organic photovoltaic cells based on ... [https://www.sciencedirect.com/science/article/abs/pii/S0927024808002961]
- 54. Organic semiconductor density of states controls the ... [https://www.nature.com/articles/ncomms5174]
- 55. Solution-processed, semitransparent organic photovoltaics ... [https://www.nature.com/articles/s41598-020-77012-2]
- 56. X-ray absorption spectroscopy [https://en.wikipedia.org/wiki/X-ray_absorption_spectroscopy]
- 57. Related Techniques — xrayabsorption.org [https://xafs.xrayabsorption.org/related_techniques.html]
- 58. Resonant inelastic X-ray scattering for studying materials ... [https://www.sciencedirect.com/science/article/pii/S2589004225010818]
- 59. RIXS, XES and XAS studies for electronic structure of rare ... [https://www.sciencedirect.com/science/article/abs/pii/S092145262100716X]
- 60. Solvent-driven spectroscopic and quantum chemical ... [https://www.nature.com/articles/s41598-025-16607-z]
- 61. Electron Spectroscopy Analysis | Research Starters [https://www.ebsco.com/research-starters/physics/electron-spectroscopy-analysis]
- 62. Understanding of Charge Transport and Non-Linear ... [https://www.sciencedirect.com/science/article/abs/pii/S1566119925000850]
- 63. Photo-modulated charge transport in single-molecule ... [https://pubs.rsc.org/en/content/articlehtml/2025/tc/d5tc01991e]
- 64. of Reduction in GaN-based... | IEEE Xplore leakage current [https://ieeexplore.ieee.org/document/7803757/]
- 65. P3HT-based visible-light organic photodetectors using PEI/PAA... [https://pubs.rsc.org/en/content/articlelanding/2019/ra/c9ra08568h]
- 66. A review on spectral data preprocessing techniques for ... [https://www.sciencedirect.com/science/article/pii/S258900422501020X]
- 67. Demonstration of angular-momentum-resolved electron ... [https://www.nature.com/articles/s41467-025-60804-3]
- 68. Machine-learning-enhanced automatic spectral ... [https://www.nature.com/articles/s42005-024-01900-6]
- 69. Electronic Spectroscopy [https://chem.libretexts.org/Bookshelves/Physical_and_Theoretical_Chemistry_Textbook_Maps/Supplemental_Modules_(Physical_and_Theoretical_Chemistry)/Spectroscopy/Electronic_Spectroscopy]
- 70. LVI: The SO line list, MARVEL analysis of experimental transition ... [https://arxiv.org/html/2312.09732v1]
- 71. - Wikipedia HITRAN [https://en.wikipedia.org/wiki/HITRAN]
- 72. Abstract EPSC-DPS2025-910 [https://meetingorganizer.copernicus.org/EPSC-DPS2025/EPSC-DPS2025-910.html]
- 73. (PDF) CDSD -296, high resolution carbon dioxide spectroscopic ... [https://www.academia.edu/126900870/CDSD_296_high_resolution_carbon_dioxide_spectroscopic_databank_Version_for_atmospheric_applications]
- 74. -296 (Carbon Dioxide CDSD Databank): Updated and... Spectroscopic [https://zenodo.org/records/17520]
- 75. sciencedirect.com/science/article/abs/pii/S0022407319300160 [https://www.sciencedirect.com/science/article/abs/pii/S0022407319300160]
- 76. ExoMolHR: A Relational Database of Empirical High ... [https://arxiv.org/html/2504.08731v2]
- 77. The update of the line positions and intensities ... [https://www.sciencedirect.com/science/article/abs/pii/S0022407321003885]
- 78. HITRANonline [https://hitran.org/]
- 79. Multifunctional interface engineering enables efficient and ... [https://www.nature.com/articles/s41467-025-60214-5]
- 80. Emerging trends in interface processing: a comparative ... [https://link.springer.com/article/10.1007/s44405-025-00013-0]
- 81. Inverted device architecture for high efficiency single-layer ... [https://www.nature.com/articles/s41467-024-48553-1]
- 82. A comprehensive review of flexible perovskite solar cells [https://www.sciencedirect.com/science/article/abs/pii/S0038092X25004128]
- 83. Inverted perovskite solar cell based on ionic salt achieves ... [https://pv-magazine-usa.com/2025/05/05/inverted-perovskite-solar-cell-based-on-ionic-salt-achieves-26-efficiency/]
- 84. Breakthrough in Organic Solar Cells: Devices Hit 15% ... [https://www.infinitypv.com/news/breakthrough-in-organic-solar-cells-devices-hit-15-efficiency-and-high-performance-for-800-hours-using-slot-die-coating]
- 85. Research Progress in Small-Molecule Donor-Polymer ... [https://www.sciencedirect.com/science/article/abs/pii/S1566119925001314]
- 86. Device stability of inverted and conventional bulk ... [https://www.sciencedirect.com/science/article/abs/pii/S1566119913003315]
- 87. SPECT/CT and PET/CT, related radiopharmaceuticals, and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10023548/]
- 88. Radiochemistry for positron emission tomography [https://www.nature.com/articles/s41467-023-36377-4]
- 89. SPECT/CT and PET/CT, related radiopharmaceuticals, and ... [https://www.sciencedirect.com/science/article/pii/S1319016422003127]
- 90. Radionuclides in Diagnostics and Therapy of Malignant ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8773826/]
- 91. PET imaging gains momentum while SPECT maintains ... [https://www.medicaldevice-network.com/analyst-comment/pet-imaging-momentum-spect-strength-nuclear-medicine/]
- 92. PET Molecular Imaging: A Holistic Review of Current ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8073681/]
- 93. A Method for Validating PET and SPECT Cameras for ... [https://jnm.snmjournals.org/content/66/2/315]
- 94. Development of a Miniaturized Nuclear Imaging Capsule for ... [https://jnm.snmjournals.org/content/66/supplement_1/251868]
- 95. Enhanced sensitivity for electron affinity measurements of ... [https://www.nature.com/articles/s41467-025-64581-x]
- 96. World's First Discovery of New Electronic State in ... [https://group.ntt/en/newsrelease/2025/07/30/250730a.html]
- 97. Presenting data in tables and charts - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4008059/]
- 98. Figures and Charts - UNC Writing Center [https://writingcenter.unc.edu/tips-and-tools/figures-and-charts/]
- 99. Text has enhanced contrast | ACT Rule | WAI [https://www.w3.org/WAI/standards-guidelines/act/rules/09o5cg/]
- 100. Elements must meet minimum color contrast ratio thresholds [https://dequeuniversity.com/rules/axe/4.8/color-contrast]