FTIR and NMR Spectroscopy: A Comprehensive Guide to Functional Group Analysis for Biomedical Research
This article provides a comprehensive guide to the qualitative analysis of functional groups using Fourier Transform Infrared (FTIR) and Nuclear Magnetic Resonance (NMR) spectroscopy, tailored for researchers and professionals in...
FTIR and NMR Spectroscopy: A Comprehensive Guide to Functional Group Analysis for Biomedical Research
Abstract
This article provides a comprehensive guide to the qualitative analysis of functional groups using Fourier Transform Infrared (FTIR) and Nuclear Magnetic Resonance (NMR) spectroscopy, tailored for researchers and professionals in drug development. It covers the foundational principles of both techniques, detailing how molecular vibrations (FTIR) and nuclear spin interactions (NMR) provide complementary structural information. The scope extends to practical methodologies, sample preparation, and advanced applications in pharmaceutical analysis, including recent integrations with machine learning for enhanced prediction accuracy. It also addresses common troubleshooting scenarios and offers a comparative analysis to guide technique selection, validating findings through hybrid approaches and case studies. The article concludes by synthesizing key takeaways and exploring future implications for clinical diagnostics and biomedical research.
Core Principles: How FTIR and NMR Unlock Molecular Structures
The elucidation of molecular structure, particularly the identification of functional groups, represents a fundamental task in chemical research and drug development. Two analytical techniques form the cornerstone of this endeavor: Fourier-Transform Infrared (FT-IR) spectroscopy and Nuclear Magnetic Resonance (NMR) spectroscopy. While both provide critical structural information, they operate on entirely different physical principles. FT-IR probes the vibrational states of molecules, providing a characteristic fingerprint of their functional groups and chemical bonds. In contrast, NMR spectroscopy investigates the magnetic properties of specific atomic nuclei, yielding detailed insights into molecular connectivity, conformation, and dynamics [1] [2]. This application note delineates the fundamental physics underlying these techniques, provides structured experimental protocols for their application in functional group research, and demonstrates their powerful synergy through a case study on automated structure elucidation. The complementary nature of these methods enables researchers to obtain a comprehensive molecular portrait, which is indispensable in fields ranging from synthetic chemistry to pharmaceutical development.
Fundamental Physical Mechanisms
FT-IR: Probing Molecular Vibrations
Fourier-Transform Infrared spectroscopy is predicated on the interaction between infrared light and the vibrational modes of chemical bonds. When a molecule is irradiated with infrared light, chemical bonds undergo vibrational excitations—such as stretching, bending, and rocking—provided the radiation's frequency matches the vibrational frequency of the bond and the vibration induces a change in the dipole moment of the molecule [1]. The resulting absorption spectrum, typically plotted as transmittance or absorbance against wavenumber (cm⁻¹), serves as a unique molecular fingerprint. Different functional groups absorb characteristic frequencies of IR radiation; for instance, carbonyl groups (C=O) exhibit strong, sharp peaks around 1700 cm⁻¹, while hydroxyl groups (O-H) show broad absorptions in the 3200-3600 cm⁻¹ range [3]. The "fingerprint region" (400-1500 cm⁻¹) contains numerous complex vibrations that are highly specific to the entire molecular structure, though often challenging to interpret manually without computational assistance [4].
NMR: Harnessing Nuclear Spin
Nuclear Magnetic Resonance spectroscopy exploits the magnetic properties of certain atomic nuclei, such as ¹H and ¹³C, which possess intrinsic spin. When placed in a strong, static magnetic field (B₀), these nuclei can adopt distinct energy states (alignment with or against the field). Irradiation with radiofrequency pulses matching the energy difference between these states (the Larmor frequency) causes nuclei to resonate, flipping their spin states [1] [2]. The precise resonance frequency of a nucleus is exquisitely sensitive to its local electronic environment—shielded by surrounding electrons—resulting in a chemical shift (measured in parts per million, ppm) that provides critical information about the types of nuclei and their chemical surroundings. Furthermore, through-bond (J-coupling) and through-space (nuclear Overhauser effect, NOE) interactions reveal connectivity and spatial proximity between atoms, enabling full structural elucidation, including stereochemistry [2].
Table 1: Core Physical Principles of FT-IR and NMR Spectroscopy
| Feature | FT-IR Spectroscopy | NMR Spectroscopy |
|---|---|---|
| Fundamental Probe | Molecular vibrations & rotations [1] | Nuclear spin states in a magnetic field [1] [2] |
| Radiation Used | Infrared light | Radiofrequency waves [1] |
| Measured Property | Absorption of IR radiation due to dipole moment changes [1] | Resonance frequency (chemical shift) of nuclei [1] |
| Primary Information | Identification of functional groups & chemical bonds [1] | Atomic connectivity, molecular conformation, dynamics [1] |
| Key Spectral Parameters | Wavenumber (cm⁻¹), absorbance/transmittance | Chemical shift (ppm), coupling constant (Hz), integration [2] |
Experimental Protocols for Functional Group Analysis
Protocol: Functional Group Identification via Integrated FT-IR and NMR
This protocol outlines a machine-learning-assisted methodology for the accurate identification of 17 functional groups by simultaneously training on FT-IR, ¹H NMR, and ¹³C NMR data, achieving a high macro-average F1 score of 0.93 [5].
Research Reagent Solutions and Materials
Table 2: Essential Materials for Spectral Analysis
| Material/Software | Specification/Function |
|---|---|
| FT-IR Spectrometer | Equipped with ATR accessory for solid/liquid samples. |
| NMR Spectrometer | High-field instrument (e.g., 400-500 MHz) for ¹H/¹³C NMR. |
| Deuterated Solvent | CDCl₃ for consistency in NMR sample preparation [5]. |
| Computational Resources | Python/R environment with machine learning libraries (e.g., TensorFlow, Scikit-learn). |
| Spectral Databases | NIST Chemistry WebBook (FT-IR); SDBS/CAS SciFinder (NMR) [5]. |
Data Collection and Preprocessing Workflow
The following diagram illustrates the integrated workflow for sample preparation, data collection, and model training for functional group prediction.
Sample Preparation & Data Collection:
- Prepare pure analyte samples. For NMR, dissolve the sample in deuterated chloroform (CDCl₃) to maintain solvent consistency, which is critical for reproducible chemical shifts [5].
- Acquire FT-IR spectrum in the range of 400–4000 cm⁻¹. Transform transmittance values to absorbance to improve model training [5].
- Acquire ¹H NMR (0–12 ppm) and ¹³C NMR (0–220 ppm) spectra on a high-field spectrometer.
Spectral Preprocessing:
- FT-IR Processing: Apply min-max normalization by dividing each absorbance value by the maximum absorbance value of the dataset. Handle missing values using linear interpolation [5].
- NMR Processing: Employ data binning to reduce dimensionality and address data sparsity. For ¹H NMR, divide the 1–12 ppm range into 12 bins of 1 ppm intervals. For ¹³C NMR, divide the 1–220 ppm range into 44 bins of 5 ppm intervals. Assign a value of 1 or 0 to each bin based on the presence or absence of a peak, ignoring intensity information for enhanced model performance [5].
Functional Group Assignment & Model Training:
- For each compound in the training set, assign binary labels (1 for presence, 0 for absence) for the 17 target functional groups using SMARTS strings for unambiguous identification [5].
- Train an Artificial Neural Network model using the integrated preprocessed spectral data (FT-IR, ¹H NMR, ¹³C NMR). Apply stratified 5-fold cross-validation to the training data (80% of the total dataset) to avoid overfitting, holding back 20% for final performance evaluation [5]. The model's output is a simultaneous prediction for all functional groups.
Protocol: Rapid Structure Elucidation from IR Spectra Using Transformer Models
This protocol leverages a modern deep-learning architecture to predict molecular structures directly from IR spectra, demonstrating the untapped potential of IR data for full structural inference [4].
Data Sourcing and Preparation:
- Simulated Data for Pretraining: Generate a large-scale dataset of IR spectra via molecular dynamics simulations using a forcefield (e.g., PCFF). This study utilized 634,585 simulated spectra from PubChem structures, limited to molecules with 6–13 heavy atoms (C, H, O, N, S, P, halogens) [4].
- Experimental Data for Fine-Tuning: Obtain experimental IR spectra from standardized databases such as the NIST IR database for model refinement and validation [4].
Model Input and Training:
- Input Representation: Provide the model with both the experimental IR spectrum and the chemical formula of the target compound. The chemical formula acts as a strong prior to constrain the chemical space for the model's search [4].
- Window Selection: Focus the model's input on the most informative spectral regions. A merged split containing the fingerprint region (400–2000 cm⁻¹) and the C-H stretching region (2800–3300 cm⁻¹) was found to yield optimal performance [4].
- Architecture and Training: Employ an autoregressive encoder-decoder transformer model. The model is trained in a sequence-to-sequence fashion to generate the corresponding molecular structure encoded as a SMILES string from the input IR spectrum and formula [4].
Comparative Analysis and Synergistic Applications
Comparative Technique Profiles
The following diagram and table summarize the complementary strengths and operational differences between FT-IR and NMR spectroscopy.
Table 3: Operational Comparison of FT-IR and NMR Spectroscopy
| Aspect | FT-IR Spectroscopy | NMR Spectroscopy |
|---|---|---|
| Sample Preparation | Minimal; suitable for solids, liquids, and gases [1] | Requires dissolution in deuterated solvents; more complex [2] |
| Analysis Speed | Very rapid (seconds to minutes) | Slower (minutes to hours) [4] |
| Sensitivity | High (microgram range) [6] | Lower (milligram range); requires larger sample amounts [2] [6] |
| Quantitative Analysis | Possible with calibration curves | Highly quantitative without extensive calibration [7] |
| Key Limitation | Cannot determine atomic connectivity or full 3D structure [1] [6] | Lower sensitivity; high instrument cost and maintenance [4] [6] |
| Ideal Use Case | Initial, rapid screening for functional groups [8] | Definitive structural elucidation and confirmation [1] |
Synergistic Application in Pharmaceutical Analysis
The combination of FT-IR and NMR is powerfully demonstrated in pharmaceutical and materials research. For instance, a hybrid correlation method analyzing 2-Hydroxy-5-nitrobenzaldehyde across FT-IR, FT-Raman, UV-vis, and NMR techniques provided a comprehensive view of its molecular structure, vibrational wavenumbers, and electronic properties. This multi-technique approach, supported by density functional theory calculations, enabled the prediction of biological activity, suggesting the compound's potential as an analeptic agent [8]. Furthermore, the integration of handheld spectroscopy tools—including portable FT-IR—in screening pharmaceutical products at international mail facilities showcases the utility of FT-IR for rapid initial identification, with results confirmed by more specific techniques like NMR, ensuring high reliability in detecting active pharmaceutical ingredients [8].
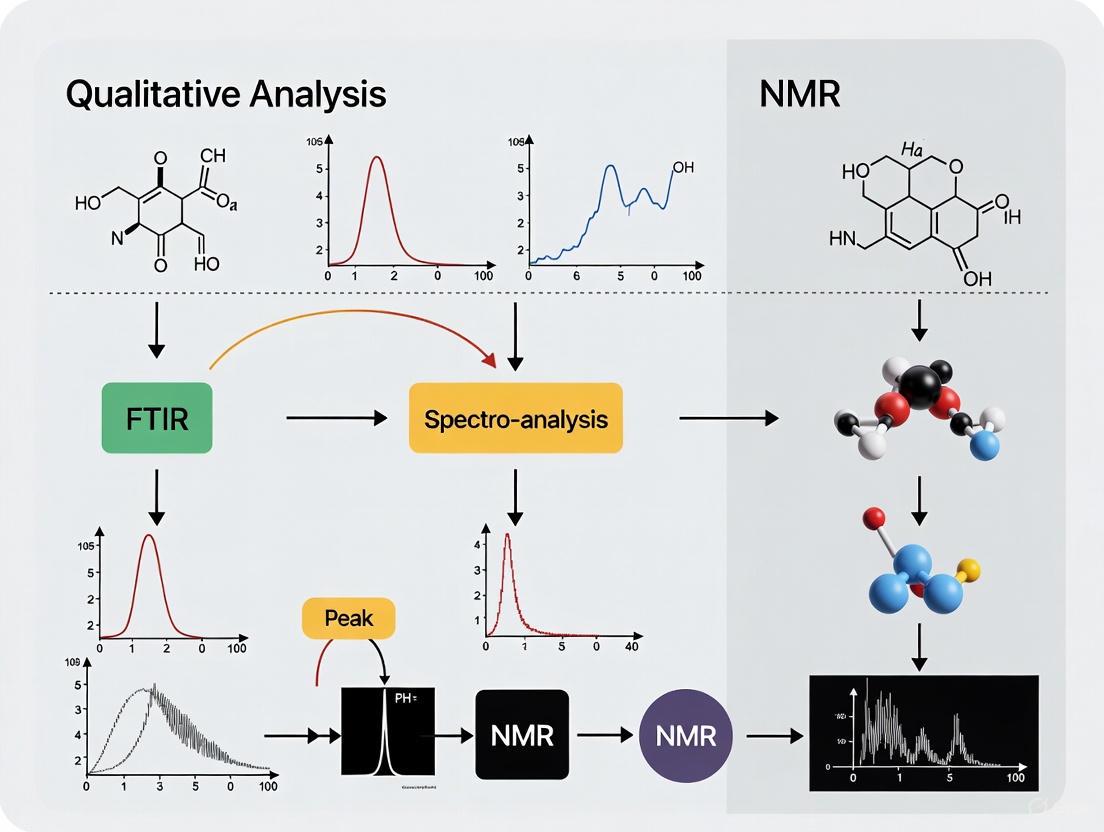
FT-IR and NMR spectroscopy, though founded on disparate physical principles—molecular vibrations versus nuclear spin—are not competing but profoundly complementary techniques. FT-IR offers unparalleled speed and simplicity for initial functional group screening, while NMR provides unparalleled detail on molecular connectivity and three-dimensional structure. As demonstrated by the advanced protocols herein, the integration of these techniques, especially when enhanced by modern machine learning algorithms, facilitates a more accurate and comprehensive analysis of molecular structures than either method could achieve alone. For researchers in drug development and related fields, leveraging this synergistic relationship is paramount for accelerating discovery and ensuring rigorous analytical outcomes.
Fourier-transform infrared (FTIR) spectroscopy is a powerful analytical technique used to obtain the infrared spectrum of absorption or emission of a solid, liquid, or gas [9]. It operates on the principle that molecules absorb specific frequencies of infrared radiation that are characteristic of their structure and chemical bonds. Unlike older dispersive spectrophotometers which measured intensity over a narrow range of wavelengths at a time, FTIR spectrometers collect high-resolution spectral data over a wide spectral range simultaneously, conferring a significant advantage in speed and accuracy [9].
The term "Fourier-transform" originates from the mathematical process required to convert the raw data (interferogram) into the actual spectrum [9]. This technique has become indispensable across numerous scientific fields including pharmaceuticals, materials science, forensics, and environmental analysis for identifying molecular compounds and functional groups [10]. When integrated into a broader analytical strategy alongside techniques like Nuclear Magnetic Resonance (NMR) spectroscopy, FTIR provides complementary data that enables comprehensive molecular characterization for functional groups research [1].
Fundamental Principles of Infrared Absorption
The Infrared Absorption Process
Infrared spectroscopy is based on the interaction between infrared light and matter, specifically exploiting the fact that molecules absorb infrared radiation at frequencies that match their natural vibrational frequencies [11]. When a molecule is exposed to infrared radiation, a portion of the incident infrared radiation is absorbed by the sample while the remainder is transmitted [12]. This absorption occurs because covalent bonds in molecules are not static but behave as if they are vibrating springs connecting atoms together [13].
For a molecule to absorb IR radiation, the vibrations or rotations within the molecule must cause a net change in the dipole moment of the molecule [12]. This dipole moment change occurs because of uneven charge distribution in chemical bonds. For example, in a carbon-oxygen bond, the difference in electronegativity between the atoms causes the electrons to be shared unevenly, leading to a partial positive charge (δ+) on the carbon and a partial negative charge (δ-) on the oxygen, creating a bond dipole [13]. When the infrared radiation frequency matches the natural vibrational frequency of the bond, energy is absorbed, and the amplitude of the vibration increases.
The ball-and-spring model of molecular vibrations provides a useful framework for understanding this process [13]. In this model, atoms are represented as balls and chemical bonds as springs. The stretching and bending of these "springs" corresponds to the absorption of infrared energy. The precise frequency at which a bond absorbs infrared radiation depends on two key factors: the strength of the bond (force constant, k) and the masses of the atoms involved (reduced mass, MR), as described by the fundamental equation [13]:
W = (1 / (2πc)) * √(k/MR)
Where W is the peak wavenumber position (cm⁻¹), c is the speed of light, k is the force constant, and MR is the reduced mass of the system. This equation explains why stronger bonds (higher k) and bonds between lighter atoms (lower MR) vibrate at higher frequencies and thus absorb at higher wavenumbers [13].
Molecular Vibrations and IR Activity
Molecules exhibit complex vibrational patterns that can be categorized into two main types: stretching vibrations (which involve changes in bond length) and bending vibrations (which involve changes in bond angle) [11]. Stretching vibrations can be further classified as symmetric or asymmetric, while bending vibrations include scissoring, rocking, wagging, and twisting motions [11].
Even simple molecules like water exhibit multiple vibrational modes: symmetric stretch, antisymmetric stretch, and deformation (bending) vibration [11]. Each of these vibrations occurs at different frequencies that are unique to the chemical bond and compound. The symmetric and antisymmetric stretches for water occur in the range of 2700 to 3700 cm⁻¹, while the deformation vibration occurs around 1650 cm⁻¹ [11].
The number of fundamental vibrations for a linear molecule is given by 3N-5, where N is the number of atoms in the molecule, while for non-linear molecules, the number is 3N-6 [13]. This means that as molecular complexity increases, the number of potential absorption bands increases accordingly, creating a unique "fingerprint" for each compound.
Figure 1: FTIR Process Workflow. This diagram illustrates the sequential process from IR source to spectral output, highlighting key stages including sample interaction, vibration excitation, and signal detection.
FTIR Instrumentation and Technology
The Michelson Interferometer
The core technological advancement that enables FTIR spectroscopy is the Michelson interferometer, which replaces the monochromator used in dispersive IR instruments [9]. In an FTIR spectrometer adapted with a Michelson interferometer, light from a polychromatic infrared source (approximately a black-body radiator) is collimated and directed to a beam splitter [9]. Ideally, 50% of the light is refracted toward a fixed mirror and 50% is transmitted toward a moving mirror [9].
The light reflects from both mirrors back to the beam splitter, and a portion of the original light passes into the sample compartment where it is focused on the sample [9]. After passing through the sample, the light is refocused onto the detector [9]. The key measurement is the difference in optical path length between the two arms of the interferometer, known as the retardation or optical path difference (OPD) [9]. An interferogram is obtained by varying the OPD and recording the signal from the detector for various values of OPD [9].
The interferogram represents a raw data signal that contains information about all infrared frequencies absorbed by the sample, but in a form that is not directly interpretable. The conversion of this raw data into a recognizable spectrum requires the application of a Fourier transform, a mathematical algorithm that deconvolutes the individual frequencies from the complex interferogram signal [9] [11].
Measurement Techniques in FTIR
Modern FTIR spectroscopy offers several measurement techniques tailored to different sample types and analytical requirements:
Transmission Spectroscopy: This is the "original" technique where IR light passes directly through the sample [11]. Samples often require preparation through dilution with non-absorbing materials like KBr for solids or CCl₄ for liquids to avoid total absorbance [11]. Transmission is widely used in FT-IR microscopy for applications in forensics and analysis of tissue samples and microplastics [11].
Attenuated Total Reflection (ATR): This has become the primary measurement technique due to minimal sample preparation requirements and non-destructive analysis [11]. The sample is placed on a crystal (typically diamond, germanium, or zinc selenide), and IR light is directed through the crystal where it interacts with the sample through an evanescent wave [11]. The light only interacts with the first few microns of the sample, producing high-quality spectra with little preparation [11].
Reflectance Spectroscopy: This technique detects IR light reflected off the surface of the sample rather than transmitted through it [11]. Variants include reflection-absorption (for thin samples on reflective substrates), specular reflection (for reflective surfaces like polymers or gemstones), and diffuse reflection (used in DRIFTS for solids like soils or catalysts) [11].
Table 1: Comparison of FTIR Sampling Techniques
| Technique | Sample Preparation | Best For | Advantages | Limitations |
|---|---|---|---|---|
| Transmission | Extensive preparation required; solids must be ground and mixed with KBr; liquids diluted with solvent | Polymer films, proteins, samples with oil in water; FT-IR microscopy | Considered the "standard" for qualitative analysis; wide applicability | Time-consuming preparation; destructive technique |
| ATR | Minimal to no preparation; sample simply placed on crystal | Virtually all sample types; particularly useful for solids, liquids, pastes | Non-destructive; rapid analysis; high-quality spectra; minimal sample prep | Slight spectral differences compared to transmission (corrected with software) |
| DRIFTS | Careful sample preparation required | Solid samples such as soils, concrete, catalysts | Excellent quantitative results for solids | More difficult to perform; requires experience |
Interpreting FTIR Spectra
Understanding the FTIR Spectrum
An FTIR spectrum is a plot that graphically represents how a sample absorbs infrared light across different frequencies [14]. The x-axis represents wavenumbers (cm⁻¹), which indicate the energy of molecular vibrations, with the typical mid-infrared range spanning from 4000 to 400 cm⁻¹ [14]. Higher wavenumbers correspond to higher energy vibrations, such as O-H and C-H stretching [14]. The y-axis represents the amount of infrared radiation absorbed or transmitted by the sample, expressed as either absorbance or transmittance [14]. In absorbance spectra, peaks represent frequencies where bonds absorb infrared light, while in transmittance spectra, these absorptions appear as downward valleys [14].
Each peak, or absorbance band, corresponds to the vibration of a specific atomic group within the molecule [14]. These vibrations occur at characteristic frequencies based on bond type, bond strength, and surrounding chemical environment [14]. For example, carbonyl (C=O) stretching typically appears as a sharp, intense peak near 1700 cm⁻¹ [14]. The position, intensity, and shape of peaks provide critical information for identifying compounds and functional groups [14].
Systematic Spectral Interpretation Approach
A structured approach to interpreting FTIR spectra ensures accurate identification of functional groups and compounds:
Verify Spectrum Quality: Begin with a high-quality spectrum exhibiting low noise, minimal baseline offset, a flat baseline, peaks on scale, and absence of spectral artifacts [15]. Common artifacts include water vapor peaks around 3400 cm⁻¹ and CO₂ peaks near 2300 cm⁻¹ [15].
Identify Key Spectral Regions: The infrared spectrum can be divided into distinct regions that correspond to different types of molecular vibrations [14]:
Figure 2: FTIR Spectral Regions. This diagram illustrates the four key regions of an FTIR spectrum and the primary molecular vibrations associated with each region.
Analyze Peak Characteristics: Examine not just peak positions but also their shape and intensity. Broad peaks between 3650 and 3250 cm⁻¹ typically indicate hydrogen bonding, commonly seen in hydroxyl (-OH) and amine (-NH) groups [14]. Sharp peaks, such as the C≡N stretch near 2200 cm⁻¹, characterize isolated or weakly interacting polar bonds [14]. Strong peaks in the carbonyl region (1850-1650 cm⁻¹) suggest highly polar bonds like ketones, aldehydes, and esters [14].
Leverage the Fingerprint Region: The region from 1500 to 500 cm⁻¹ contains complex absorption patterns unique to individual compounds [14]. While this region is often too complex for direct functional group identification, it provides a distinctive "fingerprint" for comparing unknown spectra with reference libraries to confirm compound identity [14] [10].
Utilize Reference Libraries and Software: Modern FTIR analysis is supported by comprehensive spectral databases and sophisticated software tools that facilitate automated matching and identification [14]. These resources are particularly valuable for interpreting the complex patterns in the fingerprint region [14].
Table 2: Characteristic IR Absorption Frequencies of Common Functional Groups
| Functional Group | Bond Type | Absorption Range (cm⁻¹) | Peak Characteristics |
|---|---|---|---|
| Hydroxyl | O-H stretch | 3200-3600 | Broad, strong (hydrogen-bonded); sharper (free) |
| Carbonyl | C=O stretch | 1650-1750 | Strong, sharp; exact position varies by compound type |
| Amine | N-H stretch | 3300-3500 | Medium, sharp to rounded |
| Nitrile | C≡N stretch | 2200-2260 | Medium, sharp |
| Alkyne | C≡C stretch | 2100-2260 | Variable sharpness and intensity |
| Alkyl | C-H stretch | 2850-3000 | Medium to strong |
| Methyl | C-H bend | 1370-1380 and 1450-1470 | Medium, often doublet |
| Aromatic | C=C stretch | 1500-1600 | Variable, often multiple peaks |
| Alkenyl | C=C stretch | 1620-1680 | Variable intensity |
| Ether | C-O stretch | 1000-1300 | Strong, broad |
Experimental Protocols for FTIR Analysis
Sample Preparation Methods
Proper sample preparation is critical for obtaining high-quality FTIR spectra. The appropriate method depends on the sample state and the measurement technique being employed:
ATR Sampling Protocol:
- Ensure the ATR crystal is clean by wiping with appropriate solvent (e.g., methanol) and allowing to dry completely.
- Place the sample in direct contact with the ATR crystal. For solids, apply sufficient pressure using the instrument's pressure arm to ensure good contact.
- For liquid samples, place a few drops directly onto the crystal or use a liquid cell attachment.
- Collect the spectrum without further preparation. ATR is particularly suitable for samples that are difficult to manipulate, such as gels, pastes, and irregular solids [11].
Transmission Sampling Protocol for Solids (KBr Pellet Method):
- Grind 1-2 mg of sample with 100-200 mg of dry potassium bromide (KBr) in a mortar and pestle until uniformly mixed and fine-textured.
- Transfer the mixture to a die set and apply pressure under vacuum (typically 8-10 tons for 1-2 minutes) to form a transparent pellet.
- Place the pellet in a suitable holder and position it in the sample compartment of the FTIR spectrometer.
- After analysis, clean the die set thoroughly with appropriate solvent to prevent cross-contamination [11].
Transmission Sampling Protocol for Liquids:
- Prepare a solution of the sample in a suitable solvent that does not have significant absorptions in the spectral region of interest (e.g., CCl₄, CHCl₃).
- Fill a liquid cell with a pathlength appropriate for the sample concentration (typically 0.1-1.0 mm).
- Assemble the cell, ensuring windows are clean and free of streaks.
- Place the cell in the sample compartment and collect the spectrum [11].
Instrument Operation and Data Collection
A standardized approach to instrument operation ensures reproducible and reliable results:
Instrument Preparation: Allow the FTIR spectrometer to warm up for at least 30 minutes to ensure source and detector stability. Purge the instrument with dry air or nitrogen to reduce atmospheric water vapor and CO₂ interference [15].
Background Measurement: Collect a background spectrum under identical conditions to the sample measurement but without the sample present. For ATR, this means measuring with a clean crystal; for transmission, with an empty sample holder or reference pellet [15].
Sample Measurement: Position the prepared sample and collect the spectrum using appropriate parameters (typically 4 cm⁻¹ resolution and 16-64 scans as a balance between signal-to-noise ratio and collection time) [15].
Data Processing: Apply necessary processing functions such as baseline correction, atmospheric suppression, and if using ATR, apply the ATR correction algorithm to compensate for the wavelength-dependent penetration depth [11].
Research Reagent Solutions
Table 3: Essential Materials for FTIR Analysis
| Reagent/Material | Function | Application Notes |
|---|---|---|
| Potassium Bromide (KBr) | IR-transparent matrix for solid samples | Must be dry and spectroscopic grade; forms transparent pellets under pressure |
| Diamond ATR Crystal | Internal reflection element for ATR | Chemically inert, durable; suitable for most samples |
| Zinc Selenide (ZnSe) ATR Crystal | Internal reflection element for ATR | Higher refractive index than diamond; not suitable for acidic or aqueous samples |
| Germanium ATR Crystal | Internal reflection element for ATR | High refractive index; excellent for high-index materials |
| Carbon Tetrachloride (CCl₄) | Solvent for liquid samples | IR-transparent in many regions; useful for preparing liquid samples |
| Chloroform (CHCl₃) | Solvent for liquid samples | Useful alternative to CCl₄ with different transparency windows |
| Mineral Oil (Nujol) | Mulling agent for solid samples | Suspension medium for powders; exhibits characteristic C-H absorptions |
FTIR in Pharmaceutical Research and Development
In drug development, FTIR spectroscopy serves as a critical analytical tool for multiple applications throughout the pharmaceutical pipeline. It is used to verify the chemical identity of raw materials, active pharmaceutical ingredients (APIs), and excipients, ensuring compliance with regulatory requirements [16]. FTIR can detect and quantify polymorphic forms of drug substances, which is crucial since different crystal forms can significantly impact a drug's bioavailability, stability, and processing characteristics [16].
The technique is also employed to study drug-excipient compatibility during formulation development, helping identify potential interactions that could affect drug stability or performance [16]. Additionally, FTIR is used for quality control testing of final drug products, confirming identity and detecting potential contaminants or degradation products [16]. The non-destructive nature of ATR-FTIR makes it particularly valuable for analyzing finished products without compromising their integrity.
FTIR spectroscopy frequently complements other analytical techniques in pharmaceutical analysis. While Nuclear Magnetic Resonance (NMR) spectroscopy provides detailed information about carbon skeleton connectivity, stereochemistry, and three-dimensional molecular structure [1], FTIR offers complementary data on functional groups, molecular symmetry, and specific chemical bonds [1]. This combined approach delivers a more comprehensive molecular characterization than either technique alone.
Comparative Analysis with NMR Spectroscopy
FTIR and NMR spectroscopy represent complementary approaches in functional group research, each with distinct strengths and applications:
Information Obtained: FTIR measures the absorption of infrared radiation by molecules, providing information about vibrational and rotational modes of chemical bonds, which is particularly useful for identifying functional groups and determining chemical composition [1]. In contrast, NMR measures the interaction of atomic nuclei with a magnetic field and radiofrequency radiation, providing detailed information about nuclear environments and molecular structure, making it especially valuable for elucidating atom connectivity and stereochemistry [1].
Sample Requirements: FTIR can analyze a wide range of samples including liquids, gases, and solids with minimal preparation in many cases [1]. NMR is primarily used for liquid samples and solid-state samples containing nuclei with magnetic properties (such as ^1H or ^13C), with particular strength in analyzing small organic molecules and biomolecules [1].
Structural Information Content: FTIR is most suited for obtaining information about functional groups, molecular symmetry, and chemical bonds, helping identify specific chemical groups (-OH, -C=O) and structural features (double bonds) [1]. NMR provides more detailed structural information including bond connectivity, stereochemistry, and three-dimensional arrangement of atoms in a molecule, along with dynamic information about molecular motion [1].
The complementary nature of these techniques is illustrated in research applications such as soil organic carbon analysis, where both FTIR and solid-state ^13C NMR have been used to characterize molecular composition, with studies showing improved correlation between the techniques after hydrofluoric acid treatment to remove interfering silicate minerals [17].
FTIR spectroscopy remains an indispensable technique in the analytical scientist's toolkit, providing rapid, non-destructive identification of functional groups and molecular structures through their characteristic infrared absorption patterns. The fundamental process of infrared absorption, governed by the relationship between bond properties and vibrational frequencies, creates unique spectral fingerprints that enable both qualitative identification and quantitative analysis.
The integration of FTIR with complementary techniques like NMR spectroscopy creates a powerful approach for comprehensive molecular characterization in functional groups research. While FTIR excels at identifying specific functional groups and monitoring chemical changes, NMR provides unparalleled insights into molecular connectivity and three-dimensional structure. Together, these techniques form a cornerstone of modern analytical chemistry, with particular importance in pharmaceutical development, materials science, and environmental analysis.
As FTIR technology continues to evolve with advancements in instrumentation, sampling techniques, and data analysis software, its applications continue to expand across scientific disciplines. The ongoing development of portable FTIR instruments further extends its utility to field applications and point-of-need testing, ensuring its continued relevance in both research and industrial settings.
Nuclear Magnetic Resonance (NMR) spectroscopy stands as a preeminent technique for elucidating molecular structure and probing the local magnetic environment of atomic nuclei. The fundamental parameter in these investigations is the chemical shift (δ), a dimensionless value expressed in parts per million (ppm) that reports on the localized electron density and magnetic field surrounding a nucleus [18]. This shift occurs because the applied magnetic field induces electrons to circulate, generating small local magnetic fields that shield or deshield the nucleus. The precise resonance frequency of a nucleus is therefore exquisitely sensitive to its immediate chemical environment, including bonding, molecular geometry, and intermolecular interactions [19] [20]. This application note details the core principles, methodologies, and protocols for using NMR spectroscopy to characterize local magnetic environments, with a specific focus on applications within functional group research and drug development.
Theoretical Foundations of Chemical Shifts
The chemical shift of a nucleus is primarily influenced by two key factors: the electronegativity of nearby atoms and the magnetic anisotropy of surrounding electron systems.
- Electronegativity Effects: Electronegative atoms, such as oxygen, nitrogen, and halogens, withdraw electron density through σ-bonds, reducing the electron cloud around nearby nuclei. This reduction leads to deshielding, moving the NMR signal to a higher chemical shift (downfield). These effects are roughly additive; for example, the chemical shift of a methyl group increases with the number of electronegative substituents [19].
- Magnetic Anisotropy: π-Electron systems, such as those found in aromatic rings or carbonyl groups, generate ring currents when placed in a magnetic field. This creates a magnetic field that is non-uniform in space, which can shield or deshield nearby nuclei depending on their spatial position relative to the π-system. A classic example is the 14 π-electron bridged annulene, where anisotropic effects cause proton NMR signals to appear at remarkably high fields (e.g., +22.2 ppm and +12.6 ppm) [19].
Table 1: Additive Effect of Electronegative Substituents on ^1H Chemical Shifts (δ, ppm)
| Compound/Substituent | X = Cl | X = Br | X = I | X = OR |
|---|---|---|---|---|
| CH₃X | 3.0 | 2.7 | 2.1 | 3.1 |
| CH₂X₂ | 5.3 | 5.0 | 3.9 | 4.8 |
| CHX₃ | 7.3 | 7.0 | 5.7 | 5.2 |
Table 2: Characteristic ^1H Chemical Shifts for Key Functional Groups
| Functional Group | Example Compound | Chemical Shift Range (δ, ppm) | Notes |
|---|---|---|---|
| -CH₃ (terminal) | 1-Propanol | 0.94 – 1.20 | Upfield |
| -CH₂- (adj. to -OH) | 1-Propanol | 3.582 | |
| -CH- (adj. to -OH) | 2-Propanol | 4.008 | |
| -OH (alcohol) | 1 & 2-Propanol | 2.16 – 2.26 | Concentration dependent |
| -CHO (aldehyde) | Propanal | 9.793 | Strongly downfield |
| -COOH (acid) | Propanoic Acid | 11.73 | Strongly downfield |
Advanced NMR Technologies for Enhanced Resolution
Modern NMR spectrometers, particularly high-field systems, provide significant advantages for probing subtle differences in local magnetic environments.
- High-Field NMR Spectrometers: The spectral resolution of an NMR experiment increases proportionally with the magnetic field strength (B₀). Higher magnetic fields increase the separation between the resonant frequencies of nuclei, leading to better dispersion of signals and simplifying the interpretation of complex spectra [21].
- Cryogenically Cooled Probes (Cryoprobes): A key advancement in probe technology involves the cryogenic cooling of coils and preamplifiers. This cooling significantly reduces electronic noise, thereby dramatically improving the signal-to-noise ratio (SNR). The SNR is proportional to the magnetic field strength raised to the power of 3/2. Probes like the broadband direct observe cryoprobe (DOCP) enable high-sensitivity detection of a wide range of nuclei at natural abundance, circumventing the need for costly isotope labeling [21].
Experimental Protocols
Protocol: Gas-Phase NMR for Isolated Molecule Analysis
Objective: To determine the fundamental NMR parameters (chemical shift δ₀, spin-spin coupling J₀) of a medical gas or volatile compound in the absence of solvent effects, providing benchmark data for theoretical calculations and in vivo studies [20].
Materials:
- High-Field NMR Spectrometer: Equipped with a probe suitable for the target nucleus (e.g., ¹H, ¹⁹F, ¹²⁹Xe, ¹⁵N, ¹⁷O).
- Specialized Glassware: High-pressure NMR tubes or glass ampoules for sealing gaseous samples.
- Medical Gases: High-purity analytes (e.g., N₂O, Xe, O₂ as a perturber).
- Reference Compound: Tetramethylsilane (TMS) for ¹H, ¹³C, and ²⁹Si referencing, or an isolated ³He atom for absolute shielding calibration [20].
Procedure:
- Sample Preparation: Transfer the gaseous analyte into a pre-evacuated, specialized NMR tube or glass ampoule. Ensure the sample is sealed to prevent contamination from atmospheric oxygen, which can cause paramagnetic shifts [20].
- Instrument Setup: Tune and match the NMR probe to the target nucleus. For low-density gases, a sufficient number of transients must be acquired to achieve an adequate signal-to-noise ratio.
- Data Acquisition: Acquire the NMR spectrum at low gas density to approximate the conditions of an isolated molecule. Standard pulse sequences designed for liquids are typically applicable [20].
- Data Analysis:
- Measure the chemical shift (δ₀) relative to the appropriate reference.
- Measure spin-spin coupling constants (J₀).
- To obtain the absolute shielding constant (σ₀), use the relationship: δ₀ ≈ σref – σ₀, where σref is the absolute shielding of the reference compound [20].
- Intermolecular Studies: To investigate solvent effects, introduce a controlled amount of inert gas (e.g., SF₆) as a gaseous solvent and repeat the measurements at different densities to separate the intrinsic molecular parameters from intermolecular contributions [20].
Table 3: Gas-Phase NMR Parameters for Selected Medical Gases
| Compound | Observed Nucleus | Chemical Shift δ₀ (ppm) | Absolute Shielding σ₀ (ppm) | Spin-Spin Coupling (Hz) |
|---|---|---|---|---|
| Helium-3 (³He) | ³He | 0 | 59.967 | 0 |
| Nitrous Oxide (N₂O) | ¹⁵N (terminal) | –235.3 | +99.55 | ¹JNN = –8.9 |
| Nitrous Oxide (N₂O) | ¹⁷O | –106.4 | +183.86 | - |
| Xenon (¹²⁹Xe) | ¹²⁹Xe | 0 | +6938 | 0 |
| Water (H₂¹⁷O) | ¹⁷O | –35.20 | +325.4 | ¹JOH = –78.2 |
Protocol: Investigating Prepolymerization Interactions via NMR and FTIR
Objective: To identify and characterize non-covalent interactions, such as hydrogen bonding, between a template molecule (e.g., 2-aminopyridine) and a functional monomer (e.g., methacrylic acid) in a prepolymerization solution [22].
Materials:
- NMR Spectrometer: For ¹H NMR analysis.
- FTIR Spectrometer: To complement NMR data and study temperature effects.
- Template and Monomer: e.g., 2-aminopyridine and methacrylic acid.
- Deuterated Solvent: Suitable for both NMR and the chemical system under study.
Procedure:
- Sample Preparation: Prepare solutions of the template, monomer, and their mixture in the chosen deuterated solvent.
- Job Plot Analysis (Titration):
- Prepare a series of solutions where the total molar concentration of template and monomer is constant, but their mole fractions vary.
- Acquire ¹H NMR spectra for each solution.
- Plot the chemical shift change (Δδ) of a key proton (e.g., the template's NH₂ or aromatic proton) against the mole fraction of the template.
- The maximum complexation occurs at the mole fraction corresponding to the complex's stoichiometry (e.g., a maximum at 0.5 mole fraction indicates a 1:1 complex) [22].
- FTIR Analysis: Acquire IR spectra of the individual components and the mixture. Look for shifts in characteristic bands, such as the carbonyl stretch of the acid or the N-H stretch of the aminopyridine, which indicate hydrogen bond formation. FTIR is particularly useful for observing the stabilization of these interactions at lower temperatures [22].
- Data Correlation: Correlate the band shifts observed in FTIR with the chemical shift changes observed in ¹H NMR to confirm the mechanism and strength of interaction. Compare the results with structural analogs (e.g., 3-aminopyridine) to understand the effect of template pKₐ on the interaction strength [22].
Computational NMR and Machine Learning Approaches
Interpreting NMR spectra of disordered materials, such as solid solutions, requires advanced computational models that account for local compositional fluctuations.
- Grand-Canonical Ensemble Approach: Traditional canonical ensemble models, which sample configurations at a fixed composition, often fail to capture the full range of local environments. A grand-canonical approach allows for the simulation of all possible local chemical environments around a probe nucleus, providing a more accurate and comprehensive interpretation of the experimental NMR spectrum [23] [24].
- Machine Learning (ML) for Shift Prediction: To mitigate the high computational cost of density functional theory (DFT) calculations for numerous configurations, machine learning models can be trained to predict NMR chemical shifts. Using descriptors derived from local atomic structures, ML can achieve a significant reduction in computational demand while maintaining predictive accuracy [23] [24].
- Workflow: The process involves generating a representative ensemble of local structures, computing chemical shifts (via DFT for a subset and ML for the full set), and calculating the final spectrum as a weighted average of contributions from all environments [24].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 4: Key Research Reagent Solutions for NMR Studies of Local Environments
| Item | Function/Application |
|---|---|
| High-Field NMR Spectrometer with Cryoprobe | Provides high spectral resolution and sensitivity for detecting a wide range of nuclei, crucial for complex molecules and natural abundance studies [21]. |
| Deuterated Solvents (e.g., CDCl₃, DMSO-d₆) | Provide a signal for NMR spectrometer field-frequency lock, enabling stable data acquisition. The choice of solvent can influence observed chemical shifts, particularly for protons involved in hydrogen bonding [19] [18]. |
| Internal Standard (e.g., TMS) | Provides a reference peak (δ = 0 ppm) for accurate chemical shift determination in quantitative and qualitative analysis [18]. |
| Specialized Gas NMR Glassware | High-pressure tubes and ampoules for the preparation and sealing of gaseous samples, allowing for the study of isolated molecules and intermolecular interactions in the gas phase [20]. |
| Isotope-Enriched Compounds (e.g., ¹⁷O, ¹⁵N) | Enhance the sensitivity for magnetically dilute nuclei, making NMR studies of biologically relevant atoms like oxygen and nitrogen feasible [20]. |
Workflow for NMR-Based Functional Group Analysis
The following diagram outlines a generalized workflow for probing the local magnetic environment to identify functional groups and intermolecular interactions.
NMR spectroscopy provides an unparalleled window into the local magnetic environment of atoms, offering detailed insights that are critical for functional group identification, understanding intermolecular interactions, and guiding drug development. The continuous advancement in spectrometer technology, exemplified by high-field magnets and cryoprobes, coupled with innovative computational approaches like grand-canonical sampling and machine learning, is expanding the frontiers of NMR applications. By employing the protocols and methodologies outlined in this document, researchers can leverage the full power of NMR to solve complex structural problems from isolated molecules to disordered solid-state materials.
Fourier-Transform Infrared (FTIR) spectroscopy is a cornerstone analytical technique in modern research and drug development for identifying organic compounds. Its principle is based on the absorption of infrared radiation by molecules, which causes covalent bonds to vibrate at characteristic frequencies. These frequencies provide a molecular "fingerprint" [25]. The resulting infrared spectrum is typically divided into two critical areas for interpretation: the Functional Group Region (approximately 4000–1500 cm⁻¹) and the Fingerprint Region (approximately 1500–500 cm⁻¹) [26] [27]. This application note details the characteristics of these regions and provides structured protocols for their use in qualitative analysis, framing this knowledge within the broader context of spectroscopic techniques like NMR for functional group identification.
Characteristic Spectral Regions in FTIR
The infrared spectrum is conceptually divided into distinct areas that provide different structural information. The table below summarizes the core characteristics of these two primary regions.
Table 1: Core Characteristics of the Functional Group and Fingerprint Regions in FTIR Spectroscopy.
| Feature | Functional Group Region | Fingerprint Region |
|---|---|---|
| Spectral Range | 4000 – 1500 cm⁻¹ [26] [27] | 1500 – 500 cm⁻¹ [26] [27] |
| Primary Information | Identification of specific functional groups (e.g., -OH, C=O, N-H) [28] | Confirmation of molecular structure; unique pattern for each compound [26] |
| Band Origins | Stretching vibrations of diatomic units and key functional groups [27] | Complex mix of bending, skeletal vibrations, and single-bond stretches [26] [29] |
| Interpretation | Straightforward; peaks can be directly assigned to bond types [30] | Complex; pattern-matching is required rather than individual peak assignment [26] |
| Band Appearance | Often strong, sharp, and isolated [29] | Numerous, closely spaced peaks [26] |
The following workflow outlines a strategic approach to interpreting an FTIR spectrum, leveraging the distinct information from each region.
The Functional Group Region
This high-wavenumber region contains absorptions from stretching vibrations of key bonds. The peaks here are typically strong and can often be directly correlated to the presence of specific functional groups in the molecule [28] [30]. A strategic analysis focuses on a few high-yield areas [30].
Table 2: Characteristic Absorptions in the Functional Group Region for Common Functional Groups. Data consolidated from multiple sources [31] [28] [29].
| Functional Group | Bond | Peak Position (cm⁻¹) | Peak Characteristics |
|---|---|---|---|
| Alcohol / Phenol | O-H stretch | 3200–3550 [31] [28] | Strong, broad [31] [28] |
| Carboxylic Acid | O-H stretch | 2500–3300 [31] [29] | Very strong, very broad [31] |
| Primary / Secondary Amine | N-H stretch | 3300–3500 [31] [27] | Medium, sharp (often doublet for primary) [31] [27] |
| Alkyne | ≡C-H stretch | ~3300 [31] [28] | Strong, sharp [31] |
| Alkene / Aromatic | =C-H stretch | 3000–3100 [31] [28] | Medium to weak |
| Aldehyde | C-H stretch | 2695–2830 [31] [27] | Medium (Fermi doublet) [31] |
| Alkane | C-H stretch | 2840–3000 [31] [28] | Medium |
| Carbonyl (General) | C=O stretch | 1630–1815 [31] [29] | Strong, sharp ("sword-like") [30] |
| • Ketone | C=O stretch | 1705–1725 [31] [28] | Strong, sharp |
| • Aldehyde | C=O stretch | 1720–1740 [31] [28] | Strong, sharp |
| • Ester | C=O stretch | 1735–1750 [31] [29] | Strong, sharp |
| • Carboxylic Acid | C=O stretch | 1706–1720 [31] [28] | Strong, broad |
| • Amide | C=O stretch | 1630–1690 [31] [29] | Strong, sharp |
| Nitrile | C≡N stretch | 2222–2260 [31] [32] | Weak to medium, sharp |
| Alkyne | C≡C stretch | 2100–2260 [31] [28] | Weak |
The Fingerprint Region
The fingerprint region (1500–500 cm⁻¹) arises from a complex combination of bending vibrations, skeletal vibrations (stretching and bending of the carbon backbone), and single-bond stretching vibrations (e.g., C-O, C-N) [26] [29]. While individual peaks are difficult to assign, the overall pattern is highly unique to the entire molecular structure. This region is most effectively used to confirm a compound's identity by comparing its spectrum to a reference spectrum measured under identical conditions [26]. For instance, the IR spectra of propan-1-ol and propan-2-ol, while similar in the functional group region, are completely different in the fingerprint region, allowing for their differentiation [26].
Experimental Protocol: FTIR Analysis for Functional Group Identification
Research Reagent Solutions & Essential Materials
Table 3: Essential materials and reagents for FTIR sample preparation.
| Item Name | Function / Application |
|---|---|
| FTIR Spectrometer | Instrument to generate and measure infrared absorption spectra. |
| Potassium Bromide (KBr) | Transparent to IR radiation; used for preparing solid sample pellets under high pressure [27]. |
| NaCl or AgBr Plates | Polished salt plates transparent to IR radiation; used for analyzing liquid samples as a thin film [27]. |
| Perchlorated Solvents (CCl₄, CDCl₃) | IR-transparent solvents for analyzing solid samples in solution [27]. |
| Mortar and Pestle | For grinding solid samples to a fine powder for KBr pellet preparation. |
| Hydraulic Press | To apply high pressure for forming transparent KBr pellets. |
Step-by-Step Workflow
The following protocol outlines the key steps from sample preparation to spectral interpretation.
Protocol 1: Sample Preparation and Data Acquisition
1.1 Sample Preparation (Choice of method is critical) [27]
- For Solid Samples (KBr Pellet Method):
- Grind 1–2 mg of the dry solid sample with 200–300 mg of finely powdered, anhydrous KBr in a mortar and pestle.
- Place the mixture in a die and subject it to high pressure (typically ~10 tons) under vacuum for 1–2 minutes to form a transparent pellet.
- For Liquid Samples (Thin Film Method):
- Place a drop of the neat liquid on one polished face of a salt plate (e.g., NaCl or AgBr).
- Carefully place a second salt plate on top to spread the liquid into a thin film, creating a liquid capillary film assembly. Avoid bubbles.
- For Solutions:
- Dissolve the sample in an IR-transparent solvent (e.g., CCl₄, CHCl₃) to a typical concentration of 1–10% w/v.
- Transfer the solution to a sealed liquid cell with fixed pathlength (e.g., 0.1 mm).
1.2 Data Acquisition
- Place the prepared sample into the FTIR spectrometer compartment.
- Collect a background spectrum (e.g., empty holder, pure KBr pellet, or solvent-filled cell).
- Collect the sample spectrum over a standard range of 4000–400 cm⁻¹ with a resolution of 4 cm⁻¹. Average multiple scans (e.g., 16–32) to improve the signal-to-noise ratio.
Protocol 2: Strategic Spectral Interpretation
2.1 Initial Functional Group Analysis [30]
- Examine the 3500–3200 cm⁻¹ region: Look for broad "tongue-like" peaks indicating O-H stretches (alcohols, phenols, carboxylic acids) or sharper peaks indicating N-H stretches (amines, amides).
- Examine the 1800–1650 cm⁻¹ region: Look for strong, sharp "sword-like" peaks indicating C=O stretches (ketones, aldehydes, esters, etc.).
- Check the 3000 cm⁻¹ threshold: Absorption just above 3000 cm⁻¹ suggests alkene or aromatic C-H stretches, while absorption below 3000 cm⁻¹ suggests alkane C-H stretches [28] [30].
- Check the 2250 cm⁻¹ region: Look for weak to medium peaks indicating nitriles (C≡N) or alkynes (C≡C).
2.2 Fingerprint Region Analysis and Confirmation
- Compare with reference spectra: Use the entire fingerprint region (1500–500 cm⁻¹) to compare the unknown spectrum with a library spectrum of the suspected compound. A match in this region strongly confirms the molecular identity [26].
- Look for specific patterns: While complex, certain areas can provide supporting evidence, such as aromatic substitution patterns (900–675 cm⁻¹) or C-O stretches from alcohols and esters (1260–1000 cm⁻¹) [31].
Integration with NMR in a Broader Analytical Strategy
FTIR spectroscopy excels at rapidly identifying specific functional groups, particularly carbonyls and hydroxyls, but it is often used in conjunction with Nuclear Magnetic Resonance (NMR) spectroscopy for comprehensive structural elucidation. While FTIR provides excellent functional group intelligence, NMR is superior for determining carbon connectivity and the complete molecular skeleton [30].
Modern approaches are now leveraging machine learning (ML) to enhance the accuracy and speed of functional group identification. Recent research demonstrates that training artificial neural networks on combined FTIR and NMR (¹H and ¹³C) spectral data outperforms models using any single technique, achieving a high macro-average F1 score of 0.93 for identifying 17 different functional groups [5]. This multi-technique ML approach mirrors the expert practice of simultaneously analyzing multiple spectra and holds significant promise for accelerating research in drug development and other fields.
Nuclear Magnetic Resonance (NMR) spectroscopy stands as one of the most powerful analytical techniques for determining the structure of organic compounds [33]. Of all spectroscopic methods, it is the only one for which a complete analysis and interpretation of the entire spectrum is normally expected, providing unparalleled insight into molecular structure [33]. The chemical shift phenomenon forms the very foundation of NMR interpretation, serving as a sensitive probe of a nucleus's local electronic environment. Unlike infrared and UV-visible spectroscopy where absorption peaks are uniquely located by frequency or wavelength, NMR resonance signals depend on both the external magnetic field strength and the RF frequency, necessitating a standardized referencing system [33].
The chemical shift (δ) refers to the relative position of proton peaks on the horizontal axis of an NMR spectrum, expressed in parts per million (ppm) relative to a standard compound [34]. This relative positioning arises from the varying degrees of nuclear shielding experienced by different protons within a molecule. When placed in an external magnetic field, electrons surrounding nuclei circulate and generate induced magnetic fields that oppose the applied field, effectively shielding the nucleus from the full strength of the external magnet [33]. The extent of this shielding depends critically on the local electron density around each nucleus, making chemical shifts exquisitely sensitive to molecular structure and functional groups.
Fundamental Principles of Chemical Shifts
Theoretical Foundation
The NMR phenomenon originates from the intrinsic magnetic properties of certain atomic nuclei. Nuclei with non-zero spin (I ≠ 0), such as ¹H, ¹³C, ¹⁹F, and ³¹P, possess a magnetic moment and can exist in different energy states when placed in an external magnetic field [35] [33]. For spin-½ nuclei, two spin states exist: +½ (aligned with the field) and -½ (opposed to the field). The energy difference between these states is proportional to the external magnetic field strength and the magnetic moment of the nucleus [33].
The resonant frequency of a nucleus is influenced by its electronic environment through a phenomenon known as nuclear shielding. Electrons surrounding the nucleus circulate in the applied magnetic field, generating secondary magnetic fields that either oppose or augment the external field at the nucleus [33]. This electron shielding means that nuclei in different chemical environments require different field strengths (or frequencies) to achieve resonance, giving rise to the chemical shifts observed in NMR spectra.
Reference Standard and Chemical Shift Calculation
Tetramethylsilane (TMS) serves as the universal reference compound for both proton and carbon NMR spectroscopy, with its signal set exactly at 0 ppm [36] [37] [38]. TMS is ideal for this purpose because it is chemically inert, volatile for easy sample recovery, and produces a single sharp resonance signal that does not interfere with most organic compounds [33].
The chemical shift (δ) in parts per million is calculated using the formula:
δ = (νsample - νreference) / ν_spectrometer × 10⁶
where νsample is the resonance frequency of the sample signal, νreference is the resonance frequency of the reference signal (TMS), and ν_spectrometer is the operating frequency of the spectrometer [35]. This standardization makes chemical shifts independent of the magnetic field strength, allowing for consistent comparison of NMR data across different instruments [39].
Chemical Shift Reference Tables
The chemical shifts of protons in different functional groups fall into characteristic ranges that provide crucial structural information. The table below summarizes the typical chemical shift values for common organic functional groups, serving as a fundamental reference for spectral interpretation.
Table 1: Characteristic ¹H NMR Chemical Shifts for Common Functional Groups
| Hydrogen Type | Chemical Shift Range (ppm) | Hydrogen Type | Chemical Shift Range (ppm) |
|---|---|---|---|
| RCH₃ (1° alkane) | 0.9 - 1.0 | ROH (alcohol) | 1 - 5 |
| RCH₂R (2° alkane) | 1.2 - 1.7 | RCH=CHR (alkene) | 4.5 - 7.5 |
| R₃CH (3° alkane) | 1.5 - 2.0 | ArH (aromatic) | 6.0 - 8.7 |
| R₂NCH₃ (amine) | 2.3 - 3.0 | RCHO (aldehyde) | 9.5 - 10.0 |
| ArCH₃ (aryl methyl) | 2.2 - 2.4 | RCOOH (carboxylic acid) | 10 - 13 |
| ROCH₃ (ether) | 3.7 - 3.9 | RNH₂ (amine) | 1 - 3 |
The energy axis in an NMR spectrum is called the delta (δ) axis, with units in parts per million (ppm) [39]. The spectrum is conventionally displayed with the right side representing the low-energy region (upfield) where shielded protons resonate, and the left side representing the high-energy region (downfield) where deshielded protons resonate [39]. This orientation corresponds to increasing magnetic field strength from left to right, with the reference TMS signal at 0 ppm [33].
Structural Factors Influencing Chemical Shifts
Electronegativity Effects
The primary factor influencing chemical shifts is the electronegativity of atoms adjacent to the proton of interest. Electronegative atoms such as oxygen, nitrogen, and halogens withdraw electron density through inductive effects, reducing the electron density around nearby protons [36] [39]. This reduced electron density results in less shielding (deshielding), forcing the resonance to higher ppm values [39] [34].
The deshielding effect follows predictable patterns:
- The more electronegative the atom, the greater the downfield shift
- The effect decreases with increasing distance from the electronegative atom
- Multiple electronegative atoms have cumulative deshielding effects
This phenomenon explains why protons on carbon atoms bonded to oxygen or nitrogen resonate significantly downfield from typical alkane protons [39].
Hybridization and Magnetic Anisotropy
The hybridization state of the carbon atom to which a proton is attached significantly affects its chemical shift. sp² hybridized carbons are more electronegative than sp³ carbons due to their higher s-character (33% vs 25% s), pulling electrons closer to the carbon nucleus and deshielding attached protons [39].
Magnetic anisotropy represents another crucial factor, particularly in unsaturated systems. When molecules with π electrons are placed in a magnetic field, the circulating π electrons generate local magnetic fields that are non-uniform in space [39]. These anisotropic fields can either shield or deshield protons depending on their position relative to the π system:
- Alkenes: Protons in the plane of the double bond experience deshielding, resonating at 4-6 ppm [39]
- Aromatic systems: The ring current in benzene and other aromatics creates a strong deshielding region in the plane of the ring, shifting aromatic proton resonances to 6-9 ppm [39]
- Alkynes: Surprisingly, acetylenic protons resonate at relatively low fields (2-3 ppm) because the induced magnetic field of the triple bond's π electrons opposes the applied field in the region of the proton, creating a shielded environment [39]
Table 2: Chemical Shift Ranges by Hybridization and Functional Group
| Category | Proton Type | Chemical Shift Range (ppm) | Primary Influencing Factors |
|---|---|---|---|
| sp³ Hybridized | RCH₃ | 0.9 - 1.0 | Substitution level (1°, 2°, 3°) |
| RCH₂R | 1.2 - 1.7 | Presence of electronegative groups | |
| R₃CH | 1.5 - 2.0 | Steric effects | |
| sp² Hybridized | Alkenes (C=CH) | 4.5 - 7.5 | Magnetic anisotropy, conjugation |
| Aromatics (ArH) | 6.0 - 8.7 | Ring current, substituents | |
| Aldehydes (RCHO) | 9.5 - 10.0 | Electronegativity, anisotropy | |
| Heteroatom-Bound | Alcohols (ROH) | 1 - 5 | Hydrogen bonding, concentration |
| Amines (RNH₂) | 1 - 3 | Hydrogen bonding, pH | |
| Ethers (ROCH₃) | 3.7 - 3.9 | Electronegativity of oxygen |
Hydrogen Bonding and Exchange Effects
Protons attached to heteroatoms (O-H, N-H) exhibit unique behavior in NMR spectra. These protons experience hydrogen bonding, which reduces electron density around the proton, causing variable downfield shifts depending on the strength and extent of hydrogen bonding [35] [39]. Consequently, these signals often appear as broad peaks that can be challenging to interpret [34].
Exchangeable protons typically:
- Appear over broad chemical shift ranges (e.g., O-H from 1-5 ppm)
- Show concentration-dependent chemical shifts
- Can be broadened or eliminated by D₂O exchange
- May not show typical coupling patterns due to chemical exchange
A useful diagnostic technique involves adding a drop of deuterated water (D₂O) to the sample, which causes the disappearance of exchangeable proton signals (O-H, N-H) through H-D exchange, simplifying spectral interpretation [39] [34].
Step-by-Step Protocol for NMR Spectral Interpretation
Systematic Approach to Structure Elucidation
Interpreting NMR spectra effectively requires a structured methodology. The following workflow provides a systematic approach for extracting structural information from ¹H NMR spectra.
Chemical Shift Analysis Protocol
Objective: Identify potential functional groups present in the molecule based on chemical shift values.
Procedure:
- Survey the spectrum from 0-14 ppm, noting the number of distinct signal groups
- Identify shielded regions (0-3 ppm): Look for aliphatic chains, alkyl substitutions
- Analyze the 3-5 ppm region: Identify protons on carbons bonded to heteroatoms (O, N)
- Examine the aromatic region (6-9 ppm): Note number and pattern of aromatic signals
- Check for characteristic peaks: Aldehydes (9-10 ppm), carboxylic acids (10-13 ppm)
- Note any broad signals: Possibly exchangeable protons (O-H, N-H)
Interpretation Guidelines:
- Use Table 1 as a reference for functional group identification
- Consider cumulative effects of multiple electronegative atoms
- Note that electron-donating groups shield ortho/para aromatic protons
- Electron-withdrawing groups deshield ortho/para aromatic protons
Integration and Multiplicity Analysis
Integration Analysis: The area under each NMR peak is proportional to the number of equivalent hydrogen atoms giving rise to that signal [34]. Modern spectrometers provide digital integration values.
Protocol:
- Normalize integration values to the smallest common factor
- Convert to whole number ratios representing proton counts
- Match integration ratios to structural elements (e.g., 3:2:1 for CH₃:CH₂:CH)
Multiplicity Analysis: Splitting patterns provide information about the number of neighboring protons through the N+1 rule, where N is the number of equivalent adjacent protons [37] [34].
Common Splitting Patterns:
- Singlet (s): No neighboring protons (e.g., TMS, isolated methyl groups)
- Doublet (d): One neighboring proton
- Triplet (t): Two equivalent neighboring protons
- Quartet (q): Three equivalent neighboring protons
- Multiplet (m): Complex splitting from non-equivalent neighbors
The coupling constant (J), measured in Hz, represents the distance between peaks in a multiplet and provides information about the stereochemical relationship between coupling protons [37].
Advanced Applications in Drug Development
Quantitative NMR (qNMR) for Purity Assessment
Quantitative NMR has emerged as a powerful technique for determining compound purity and concentration in pharmaceutical development [40]. The methodology relies on the direct proportionality between NMR signal intensity and the number of nuclei generating the signal.
Internal Standard Protocol:
- Precisely weigh the sample and a reference standard of known purity
- Co-dissolve both compounds in a deuterated solvent
- Acquire NMR spectrum with sufficient relaxation delay (typically 5×T₁)
- Select well-resolved peaks for both sample and reference
- Calculate purity using the formula:
Psample = (Isample/Iref) × (Nref/Nsample) × (Msample/Mref) × (mref/msample) × Pref
Where:
- I = Integral value
- N = Number of protons represented by the peak
- M = Molecular weight
- m = Mass
- P = Purity
This method has been validated according to USP specifications, demonstrating accuracy of 98-102% and repeatability of ≤1% for drug substance analysis [40].
Mixture Analysis and Metabolite Identification
NMR spectroscopy excels at analyzing complex mixtures without requiring separation, making it invaluable for studying metabolic profiles, reaction mixtures, and formulated products.
Tequila Analysis Example:
- The ethanol methyl proton appears at 1.2 ppm (3H)
- Overlapping signals from water and ethanol CH₂ appear between 2.7-6.0 ppm
- Ethanol concentration is calculated using: EtOH = I₁/3 (where I₁ is the methyl integral)
- Water concentration is derived from: H₂O = (I₂ - 3EtOH)/2 (where I₂ is the overlapping integral)
- Volume percent ethanol is calculated by converting molar fractions using molecular weights and densities [40]
This approach demonstrated 36% ethanol content in a tequila sample compared to 38% labeled value, showcasing the accuracy of NMR for quantitative mixture analysis [40].
The Scientist's Toolkit: Essential NMR Reagents and Materials
Table 3: Essential Research Reagents for NMR Spectroscopy
| Reagent/Material | Function | Application Notes |
|---|---|---|
| Deuterated Solvents (CDCl₃, DMSO-d₆, D₂O) | Provide signal lock for spectrometer; minimize solvent interference | Choose based on compound solubility; store under anhydrous conditions |
| Tetramethylsilane (TMS) | Internal chemical shift reference (0 ppm) | Add in small quantities (~1%); volatile for easy sample recovery |
| Maleic Acid | qNMR reference standard | High purity; sharp singlet at ~6.3 ppm; stable and non-hygroscopic |
| Deuterated Water (D₂O) | Identify exchangeable protons | Add dropwise to sample; causes disappearance of O-H/N-H signals |
| NMR Tubes (5 mm standard) | Contain sample in magnetic field | High-quality tubes minimize magnetic field distortions; clean thoroughly between uses |
| Chemical Shift Databases (nmrshiftdb2) | Reference for predicted/experimental shifts | Database contains >170,000 experimental shifts with DFT validation [41] |
Experimental Workflow for NMR Sample Preparation and Analysis
The following diagram illustrates the complete workflow from sample preparation to spectral interpretation, incorporating key quality control checkpoints to ensure reliable results.
Chemical shift interpretation forms the cornerstone of NMR spectroscopy, providing a powerful lens through which to view molecular structure. The systematic approach outlined in this application note—combining chemical shift analysis, integration data, and splitting patterns—enables researchers to extract detailed structural information from NMR spectra. For drug development professionals, these techniques support critical activities from compound identification and purity assessment to metabolite profiling and mixture analysis.
The future of NMR chemical shift interpretation continues to evolve with advances in prediction algorithms like CASCADE-2.0, which now achieves sub-ppm accuracy for ¹³C NMR shift prediction using deep learning models trained on over 170,000 experimental shifts [42]. These computational tools, combined with the fundamental principles described herein, will further enhance our ability to decipher molecular structure from NMR data, accelerating research in pharmaceutical development and beyond.
From Theory to Bench: Practical Protocols and Research Applications
Within the framework of functional group research, the accuracy of qualitative analysis using Fourier Transform Infrared (FTIR) and Nuclear Magnetic Resonance (NMR) spectroscopy is fundamentally dependent on proper sample preparation. Inadequate techniques can introduce artifacts, mask critical spectral features, or lead to misinterpretation of molecular structure. This application note details standardized protocols for three foundational preparation methods: Attenuated Total Reflectance (ATR), KBr pellet formation, and NMR solvent selection. These procedures are essential for researchers and drug development professionals seeking to obtain reliable, reproducible spectroscopic data for structural elucidation.
Attenuated Total Reflectance (ATR) FTIR
Principles and Applications
ATR-FTIR spectroscopy has revolutionized the analysis of solids, liquids, and polymers by minimizing extensive sample preparation. The technique operates on the principle of generating an evanescent wave that penetrates a few micrometers into a sample in intimate contact with an internal reflection element (IRE), or crystal [43]. This method is particularly valuable for analyzing polymer laminates, fragile materials, and samples that are difficult to manipulate, as it often requires minimal pre-processing compared to transmission FTIR [43]. Its utility in functional group analysis stems from its ability to provide high-quality spectra rapidly, with enhanced spatial resolution compared to transmission modes [43].
Experimental Protocol: Micro ATR FT-IR Imaging of Polymer Laminates
Principle: This protocol utilizes a "live micro ATR imaging" feature with a focal plane array (FPA) detector to enable ultralow-pressure contact, preventing structural damage to delicate cross-sections and eliminating the need for resin embedding [43].
Workflow: The following diagram illustrates the streamlined, five-step sample preparation process for polymer laminates.
Materials and Equipment:
- FT-IR spectrometer coupled to an FT-IR microscope with a 2D FPA detector [43].
- "Slide-on" micro Germanium (Ge) ATR accessory attached to a 15× IR objective [43].
- Micro-vice sample holder [43].
- Razor blade for cross-sectioning.
Procedure:
- Sample Mounting: A small piece of the laminate is secured vertically in a micro-vice [43].
- Cross-Sectioning: The exposed edge of the sample is carefully cross-sectioned using a razor blade to create a fresh, flat surface for analysis [43].
- Instrument Setup: The micro-vice is placed on the microscope stage. The "live ATR imaging" mode, which enhances chemical contrast in real-time, is activated [43].
- Data Collection: The stage is raised slowly until the live feed indicates complete and even contact between the sample cross-section and the ATR crystal. This requires minimal applied pressure. Data is collected at 4 cm⁻¹ spectral resolution with 64 coadded scans [43].
Advantages for Functional Group Analysis:
- Rapid Analysis: Eliminates overnight resin curing, reducing preparation time from hours to minutes [43].
- Non-Destructive: The ultralow pressure prevents sample indentation, preserving samples for further analysis [43].
- High Spatial Resolution: Essential for identifying thin adhesive layers (tie-layers) within polymer laminates, as it provides a factor of four spatial resolution enhancement over transmission mode [43].
KBr Pellet Method for FTIR
Principles and Applications
The potassium bromide (KBr) pellet technique is a classic transmission FTIR method for analyzing solid samples. The principle involves dispersing a fine powder of the analyte within a transparent matrix of KBr halide salt [44] [45]. When subjected to high pressure, the mixture forms a solid, transparent pellet through which infrared light can pass. This method is ideal for functional group analysis of inorganic materials, pharmaceuticals, and organic solids, as it produces spectra with sharp, well-defined absorption bands [44] [46]. Its effectiveness relies on the sample being finely ground and thoroughly mixed with the KBr powder to reduce light scattering.
Experimental Protocol: Preparation of a 13 mm KBr Pellet
Principle: A mixture of sample and KBr powder is compressed under high pressure and, optionally, vacuum to form a transparent disk. The vacuum removes air and moisture, which can scatter light or introduce spectral artifacts [44].
Workflow: The procedure for making a KBr pellet using an evacuable die set is summarized below.
Materials and Equipment:
- Hydraulic laboratory press (e.g., Carver Press) capable of applying 8-10 tons of ram pressure [44] [47].
- Evacuable KBr die set (e.g., 13 mm diameter) [44].
- Agate pestle and mortar [47].
- Vacuum pump capable of reaching < 2 mm Hg pressure [44].
- Desiccator for storage [44].
Procedure:
- Preparation: Clean the die components thoroughly and ensure O-rings are undamaged. Assemble the die body on its base [44]. Preheat KBr in an oven to 100 °C to drive off moisture and work quickly as KBr is hygroscopic [45] [47].
- Sample Mixing: Grind 1-2 mg of the sample with 200-300 mg of dry KBr powder in a mortar to create a fine, homogeneous mixture. The typical sample-to-KBr ratio is 1:100 [44] [45] [47].
- Loading: Transfer the mixture into the bore of the assembled die. Tap lightly to distribute powder evenly. Insert the plunger [44].
- Pelleting: Position the die in the hydraulic press. Begin evacuation and maintain vacuum for 2-5 minutes. With continued evacuation, apply 8 tons of ram pressure for 2-3 minutes. Do not exceed 9 tons [44].
- Pellet Removal: Release pressure slowly and vent the vacuum. Disassemble the die and use the press to gently eject the transparent pellet [44].
- Storage: Clean all die parts after use and store in a desiccator. Flush parts with water, rinse with acetone, but avoid solvent contact with O-rings [44].
Troubleshooting: Common issues and their solutions are listed in the table below.
Table 1: Troubleshooting Guide for KBr Pellet Preparation
| Observed Fault | Probable Cause | Recommended Remedy |
|---|---|---|
| Cloudy or opaque pellet | Insufficient compaction pressure; uneven powder distribution; moisture | Use higher pressure; distribute powder more evenly; dry powder thoroughly [44] [45] |
| Pellet scatters energy | Sample particle size too large | Grind sample more thoroughly [44] |
| Clear pellet with white spots | Coarse grains in fine powder | Grind sample more thoroughly; consider sieving [44] |
| Pellet flakes or crumbles | Powder ground too finely | Grind for a shorter period [44] |
| Irregular, blotchy appearance | Dampness in powder | Dry powder more thoroughly [44] |
NMR Solvent Selection
Principles and Applications
Selecting an appropriate solvent is a critical step in NMR spectroscopy for functional group analysis and structure elucidation. The solvent must dissolve the analyte sufficiently and not interfere with the spectral region of interest. Most NMR solvents are deuterated to minimize the intense signal from protons that would otherwise obscure the sample's signals [48]. The choice of solvent influences spectral resolution, peak shape, and the observed chemical shifts, making it a vital consideration in experimental design.
Selection Criteria and Protocol
Key Criteria for Solvent Selection:
- Solubility: The solvent must adequately dissolve the sample to ensure a homogeneous solution. Higher solubility contributes to higher sensitivity, which is crucial for samples with limited availability [48].
- Purity and Moisture Content: Solvents must be chemically pure and free of water, as impurities introduce interfering signals. Deuterated solvents should be stored over molecular sieves and handled under dry conditions (e.g., using a nitrogen blanket) to prevent moisture absorption [48].
- Chemical Shift Interference: The residual proton signal of the deuterated solvent (e.g., CHCl₃ in CDCl₃) must not overlap with the key signals of the analyte [48].
- Viscosity: Lower viscosity solvents provide better spectral resolution due to improved molecular homogeneity [48].
- Cost: The price of deuterated solvents increases with the degree of deuteration. Cost-effectiveness is a consideration for large-scale studies [48].
Protocol for Solvent Selection and Sample Preparation:
- Assess Sample Polarity: Choose a solvent with polarity similar to the sample to ensure dissolution.
- Review Spectral Libraries: Consult known spectra to identify a solvent whose residual peaks do not overlap with the expected analyte signals.
- Prepare Sample Solution: Dissolve 5-10 mg of the compound in approximately 0.6-0.7 mL of the selected deuterated solvent [48].
- Add Internal Standard: Add a small amount of tetramethylsilane (TMS, 0.03%) as an internal chemical shift reference, if not already present in the solvent [48].
Table 2: Characteristics of Common Deuterated NMR Solvents
| Solvent | Key Advantages | Key Limitations | Residual Proton Peak (approx.) | Common Applications |
|---|---|---|---|---|
| CDCl₃ | Low cost; low chemical shift | Can form phosgene; hygroscopic | 7.26 ppm | Broad applicability for organic compounds [48] |
| DMSO-d₆ | High solubility for polar molecules; no residual ^1H peak | High boiling point; viscous; hygroscopic | 2.50 ppm | Polymers, pharmaceuticals, polar organics [48] |
| D₂O | Soluble for highly polar/ionic species | Exchanges with labile hydrogens | 4.79 ppm | Biomolecules, carbohydrates [48] |
| Acetone-d₆ | Good for moderately polar compounds | Can interfere with electron-deficient species | 2.05 ppm | General purpose [48] |
| Methanol-d₄ | Good for hydrogen-bonding studies | Exchanges with labile hydrogens | 4.87 ppm (OH), 3.31 ppm (CH₃) | Alcohols, acids [48] |
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential Materials for FTIR and NMR Sample Preparation
| Item | Function/Application |
|---|---|
| KBr Powder | IR-transparent matrix for forming solid pellets in transmission FTIR [44] [45]. |
| Agate Mortar and Pestle | Grinding and homogenizing solid samples with KBr to achieve optimal particle size [47]. |
| Hydraulic Pellet Press | Applies high, controlled pressure to form solid KBr pellets [44] [47]. |
| Evacuable Die Set | Die assembly used with a vacuum pump to remove air and moisture during pellet formation [44]. |
| ATR Crystal (e.g., Ge) | Internal reflection element in ATR-FTIR; creates evanescent wave for surface measurement [43]. |
| Deuterated Solvents (e.g., CDCl₃, DMSO-d₆) | NMR solvents that minimize spectral interference while dissolving the analyte [48]. |
| Tetramethylsilane (TMS) | Internal standard for calibrating chemical shift (0 ppm) in ^1H and ^13C NMR spectra [48]. |
| Molecular Sieves | Desiccant used to store and keep deuterated solvents dry, preventing water interference [48]. |
Mastering the sample preparation techniques of ATR, KBr pelleting, and NMR solvent selection is non-negotiable for achieving definitive functional group analysis. The choice of method is dictated by the sample's physical properties, the specific spectroscopic information required, and the need for either rapid screening or high-resolution data. Adherence to these detailed protocols ensures the generation of high-fidelity spectral data, which is the cornerstone of accurate structural elucidation in chemical research and drug development. As spectroscopic technologies advance, integrating these robust preparation methods with emerging techniques like machine learning will further empower researchers in decoding complex molecular structures [5].
Step-by-Step Workflow for Functional Group Identification
Functional group identification represents a cornerstone of molecular characterization in chemical research and drug development. The complementary analytical techniques of Fourier Transform Infrared (FTIR) spectroscopy and Nuclear Magnetic Resonance (NMR) spectroscopy provide a powerful framework for determining molecular structure through functional group analysis. FTIR spectroscopy detects characteristic bond vibrations, serving as an excellent initial screening method for identifying major functional groups present in a sample. NMR spectroscopy, particularly 1H NMR, provides detailed information about the carbon-hydrogen framework of organic molecules, enabling precise structural elucidation. When used together, these techniques provide orthogonal data streams that allow researchers to rapidly identify and confirm functional groups with high confidence. This application note details a standardized workflow integrating both methodologies, complete with experimental protocols and data interpretation guidelines suitable for research and development environments.
Theoretical Foundations
FTIR Spectroscopy Principles
Fourier Transform Infrared (FTIR) spectroscopy operates on the principle that chemical bonds vibrate at characteristic frequencies when exposed to infrared radiation. These vibrations include stretching (symmetrical and asymmetrical) and bending motions, each occurring at specific energy levels corresponding to the bond type and surrounding molecular environment. When infrared light interacts with a sample, energy is absorbed at frequencies matching these vibrational modes, producing an absorption spectrum that serves as a molecular "fingerprint." The resulting spectrum plots absorbance (or transmittance) against wavenumber (cm⁻¹), with specific regions corresponding to different bond types [46].
The critical recognition for practical analysis is that FTIR is exceptionally reliable for identifying specific functional groups like carbonyls and hydroxyls, though it is less effective for determining complete molecular structures independently. The technique's strength lies in its rapid, targeted identification of key functional groups through their characteristic absorption signatures [30].
NMR Spectroscopy Principles
Nuclear Magnetic Resonance (NMR) spectroscopy exploits the magnetic properties of certain atomic nuclei, most commonly the hydrogen-1 (¹H) isotope. When placed in a strong magnetic field, these nuclei absorb electromagnetic radiation at frequencies characteristic of their molecular environment. The resulting spectrum provides information about the chemical environment, number, and connectivity of hydrogen atoms in the molecule. Chemical shift (measured in parts per million, ppm) indicates the electronic environment of a nucleus, while coupling constants reveal connectivity through bonds. For functional group identification, 1H NMR chemical shifts provide definitive evidence for the presence of specific functional groups based on their distinctive effects on nearby protons [49].
Integrated Workflow for Functional Group Identification
The following diagram illustrates the comprehensive step-by-step process for functional group identification using FTIR and NMR spectroscopy:
Experimental Protocols
FTIR Spectroscopy Protocol
Sample Preparation
- Solid Samples: Grind 1-2 mg of sample with 100-200 mg of dry potassium bromide (KBr) using an agate mortar and pestle until homogeneous. Press the mixture in a hydraulic press at 10,000 psi for 1-2 minutes to form a transparent pellet. Ensure all equipment is clean and dry to avoid contamination.
- Liquid Samples: Place a drop of neat liquid between two potassium bromide plates or sodium chloride windows to form a thin film. Alternatively, prepare a dilute solution (0.1-1% w/v) in a suitable solvent (e.g., chloroform) and use a sealed liquid cell with appropriate path length.
- Polymeric Materials: For polymer samples, use the micro-attenuated total reflection (ATR) technique, which requires minimal sample preparation. Simply place a small piece of the material in contact with the ATR crystal and apply consistent pressure [50].
Instrumental Analysis
- Background Collection: Collect a background spectrum with the empty sample holder or clean ATR crystal.
- Sample Loading: Place the prepared sample in the instrument sample compartment.
- Data Acquisition: Acquire spectrum over 4000-400 cm⁻¹ range with 4 cm⁻¹ resolution and 32 scans to ensure adequate signal-to-noise ratio.
- Data Processing: Apply baseline correction and atmospheric suppression algorithms as needed.
Data Interpretation Protocol
Adopt a systematic approach to interpreting FTIR spectra by focusing on two high-priority regions first, then examining secondary regions for confirmation:
Table 1: Key FTIR Absorption Regions for Common Functional Groups
| Functional Group | Bond Type | Absorption Range (cm⁻¹) | Peak Characteristics |
|---|---|---|---|
| Hydroxyl | O-H stretch | 3200-3600 | Broad, rounded ("tongue") |
| Carbonyl | C=O stretch | 1630-1850 | Sharp, strong ("sword") |
| Amine | N-H stretch | 3200-3500 | Sharp to medium, may be doublet |
| Alkene | C-H stretch | >3000 | Medium sharpness |
| Alkane | C-H stretch | <3000 | Multiple sharp peaks |
| Nitrile | C≡N stretch | 2200-2280 | Sharp, medium intensity |
| Alkyne | C≡C stretch | 2100-2260 | Sharp, variable intensity |
Source: Adapted from [30]
NMR Spectroscopy Protocol
Sample Preparation
- Solvent Selection: Choose an appropriate deuterated solvent (e.g., CDCl₃, DMSO-d₆, D₂O) that dissolves the sample completely and does not interfere with signals of interest. For full functional group analysis, CDCl₃ is generally preferred due to its minimal residual proton signals.
- Sample Concentration: Dissolve 1-10 mg of sample in 0.5-0.7 mL of deuterated solvent to achieve optimal concentration. Overly concentrated samples can cause signal broadening, while overly dilute samples yield poor signal-to-noise ratios.
- Reference Standard: Add a small amount of tetramethylsilane (TMS) as an internal chemical shift reference (0.00 ppm) or use the residual solvent peak as a reference.
- Tube Preparation: Filter the solution if necessary to remove particulate matter, then transfer to a clean, dry NMR tube, filling to the appropriate height (typically 4-5 cm).
Instrumental Analysis
- Instrument Setup: Tune and match the probe to the sample, lock the magnetic field to the deuterium signal of the solvent, and shim the magnet to optimize field homogeneity.
- Pulse Calibration: Determine the 90° pulse length for the specific sample and probe combination.
- Spectral Acquisition: Acquire 1H NMR spectrum with the following typical parameters: spectral width of 12-16 ppm, acquisition time of 2-4 seconds, relaxation delay of 1-5 seconds, and 16-64 scans to ensure adequate signal-to-noise.
- Data Processing: Apply Fourier transformation, phase correction, and baseline correction. Reference the spectrum to TMS (0.00 ppm) or the appropriate solvent peak.
Data Interpretation Protocol
The DP4* probability method represents an advanced approach for structure verification when comparing candidate structures. This method automatically excludes outlying chemical shifts, particularly addressing the unpredictability of exchangeable protons, improving upon the original DP4 algorithm [49].
Table 2: Characteristic 1H NMR Chemical Shifts for Common Functional Groups
| Functional Group | Proton Type | Chemical Shift Range (ppm) | Characteristic Features |
|---|---|---|---|
| Alkyl | R-CH₃ | 0.7-1.3 | Often triplet or doublet |
| Alcohol/Phenol | R-OH | 1.0-5.0 (variable) | Broad, exchangeable |
| Amine | R-NH₂ | 1.0-5.0 (variable) | Broad, exchangeable |
| Alkene | =C-H | 4.5-6.5 | Various multiplicity |
| Aldehyde | R-CHO | 9.0-10.0 | Distinctive downfield |
| Carboxylic Acid | R-COOH | 11.0-12.0 | Very broad, downfield |
| Aromatic | Ar-H | 6.5-8.5 | Complex patterns |
Complementary Data Integration
Sequential Analysis Strategy
The most effective functional group identification employs a sequential strategy where FTIR provides initial functional group hypotheses that NMR subsequently refines or confirms. For example:
- FTIR indicates carbonyl presence: A strong, sharp peak at 1700-1750 cm⁻¹ suggests a carbonyl functional group.
- FTIR further characterizes carbonyl type: Additional features may suggest specific carbonyl types - esters typically show two strong C-O stretches at 1150-1300 cm⁻¹, while carboxylic acids display a broad O-H stretch at 2500-3500 cm⁻¹.
- NMR confirms and differentiates: 1H NMR chemical shifts provide definitive evidence - aldehydes show characteristic protons at 9-10 ppm, while esters lack these but may show alkoxy group protons.
- Combined confidence: Agreement between both techniques provides high-confidence identification [30] [49].
Statistical Verification
For challenging structural determinations involving similar isomers, combining NMR and IR data significantly improves verification rates. Recent studies demonstrate that at a 90% true positive rate, using NMR and IR together reduces unsolved pairs to 0-15%, compared to 27-49% using either technique alone. At a 95% true positive rate, combined methodology reduces unsolved pairs to 15-30% from 39-70% with individual techniques [49].
Table 3: Performance Comparison of Structure Verification Methods
| Method | True Positive Rate | Unsolved Pairs | Key Advantage |
|---|---|---|---|
| 1H NMR alone | 90% | 27-49% | Detailed proton environment |
| IR alone | 90% | 27-49% | Specific functional group vibrations |
| Combined NMR & IR | 90% | 0-15% | Complementary information |
| 1H NMR alone | 95% | 39-70% | Higher confidence in proton data |
| IR alone | 95% | 39-70% | Higher confidence in functional groups |
| Combined NMR & IR | 95% | 15-30% | Maximum structural confirmation |
Source: Adapted from [49]
Research Reagent Solutions
Table 4: Essential Materials for Functional Group Analysis
| Reagent/Material | Function | Application Notes |
|---|---|---|
| Potassium Bromide (KBr) | IR matrix material | Optical grade, dry; for pellet preparation |
| Deuterated Chloroform (CDCl₃) | NMR solvent | For non-polar to moderately polar compounds |
| Deuterated DMSO (DMSO-d₆) | NMR solvent | For polar compounds, enhances solubility |
| Tetramethylsilane (TMS) | NMR chemical shift reference | Internal standard (0.00 ppm) |
| Sodium Chloride (NaCl) plates | IR sample windows | For liquid samples, hygroscopic |
| Agate Mortar and Pestle | Sample preparation | For grinding solid samples with KBr |
| NMR Tubes | Sample containment | High-quality, matched for consistent results |
| ATR Crystal (Diamond) | IR sampling | Minimal preparation required |
Case Study: Poly(imino sulfone) Characterization
A practical application of this workflow is illustrated in the characterization of the novel stimulus-responsive polymer poly(imino sulfone) (PIS). FTIR analysis confirmed the presence of key functional groups through characteristic absorptions: strong carbonyl stretches appeared at approximately 1700 cm⁻¹, while N-H stretches were observed around 3300 cm⁻¹. Subsequent 1H NMR analysis provided detailed structural confirmation, showing characteristic proton environments consistent with the proposed polymer structure. This complementary approach enabled complete structural verification and functional group analysis, facilitating the material's application in advanced anti-counterfeiting and encryption technologies [51].
The integrated FTIR and NMR workflow presented herein provides a robust, standardized methodology for functional group identification that leverages the complementary strengths of both techniques. FTIR offers rapid screening and specific functional group targeting, while NMR delivers detailed structural information about the molecular framework. This orthogonal approach significantly enhances verification confidence, particularly for challenging structural determinations involving similar isomers. The systematic protocols, data interpretation guidelines, and performance metrics outlined in this application note provide researchers with a comprehensive framework for efficient and accurate functional group analysis in drug development and chemical research.
Fourier Transform Infrared (FTIR) spectroscopy and Nuclear Magnetic Resonance (NMR) spectroscopy represent cornerstone analytical techniques in modern pharmaceutical research and development. These methods provide powerful capabilities for molecular fingerprinting, enabling researchers to identify functional groups, quantify active pharmaceutical ingredients (APIs), monitor polymorphic conversions, and ensure product quality from formulation development to final quality control. Within the framework of qualitative analysis techniques for functional group research, FTIR and NMR offer complementary strengths—FTIR provides rapid information about specific bond vibrations and functional groups, while NMR delivers detailed structural insights about atomic connectivity and molecular environment. The pharmaceutical industry increasingly relies on these techniques under Quality by Design (QbD) principles and Process Analytical Technology (PAT) initiatives to enhance product quality, accelerate development timelines, and ensure regulatory compliance [52] [53].
This application note presents detailed case studies and experimental protocols demonstrating the practical application of FTIR and NMR spectroscopy in addressing real-world challenges throughout the pharmaceutical development lifecycle. By integrating these analytical techniques with advanced computational approaches, researchers can achieve unprecedented insights into drug composition, stability, and quality attributes.
FTIR Spectroscopy in Pharmaceutical Analysis
Fundamental Principles and Technical Advantages
FTIR spectroscopy characterizes molecules based on their absorption of infrared light in the mid-IR range (typically 4,000–400 cm⁻¹), generating a unique spectral "fingerprint" that reflects the vibrational modes of chemical bonds within the sample [52] [53]. The resulting spectra are highly sensitive to molecular environment, making FTIR ideal for detecting subtle changes in polymorphic forms, molecular interactions, and functional group characteristics [52].
Key advantages of FTIR spectroscopy include:
- Non-destructive analysis preserving sample integrity
- Minimal sample preparation requirements for most applications
- Rapid data acquisition enabling real-time process monitoring
- High specificity for functional group identification
- Versatility across sample types (solids, liquids, semi-solids)
The technique operates on the principle that molecules must undergo a net change in dipole moment to absorb IR radiation, making it particularly sensitive to polar functional groups [54]. Different sampling modes including attenuated total reflectance (ATR), transmission, and diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) provide flexibility for various pharmaceutical sample types [52].
Experimental Design and Methodology
Sample Preparation Protocols
Solid Dosage Forms (ATR-FTIR):
- Sample Presentation: Place solid samples (powders, tablets) directly onto the ATR crystal
- Compression: Apply consistent pressure to ensure adequate crystal contact
- Background Measurement: Collect background spectrum with clean crystal
- Data Acquisition: Typically 16-64 scans at 4 cm⁻¹ resolution
- Replication: Analyze at least three independent samples for statistical significance
Liquid Formulations (Transmission FTIR):
- Cell Assembly: Utilize liquid cell apparatus with appropriate windows (e.g., CaF₂)
- Sample Loading: Inject sufficient sample volume to avoid air bubbles
- Pathlength Selection: Choose appropriate pathlength (typically 0.1-1.0 mm) based on analyte concentration
- Equilibration: Allow 10-15 minutes for temperature equilibration and purging of atmospheric water vapor
- Background Correction: Collect background spectrum with matching empty cell or solvent reference [54]
Semi-solid Formulations (ATR-FTIR):
- Homogenization: Ensure sample uniformity before analysis
- Direct Application: Apply thin, consistent layer to ATR crystal
- Cleaning Protocol: Clean crystal thoroughly between samples with appropriate solvents
Case Study: Quantitative Analysis of Azodicarboxamide API
Experimental Protocol
Objective: Develop and validate an FTIR method for qualitative and quantitative analysis of azodicarboxamide and its potential impurities in raw materials and dosage forms [55].
Materials and Equipment:
- FTIR spectrometer with mercury-cadmium-telluride (MCT) detector
- Dimethyl sulfoxide (DMSO) for sample dissolution
- Microboat circle cell with ZnSe crystal rod (13 μm pathlength)
- Chloroform and tetrachlorocarbon for impurity analysis
Methodology:
- Standard Preparation: Prepare azodicarboxamide standards in DMSO (10-40 mg/mL)
- Sample Preparation: Dissolve raw material or extracted dosage forms in DMSO
- Spectral Acquisition: Collect spectra from 2000-1500 cm⁻¹ region (characteristic absorption region)
- Data Processing: Apply baseline correction and peak integration
- Quantification: Construct calibration curve from standard absorbance values
- Impurity Analysis: Extract potential impurities (diethylazodicarboxylate, 1,2-dicarbethoxyhydrazine, ethyl formate) in non-polar solvents for separate analysis
Table 1: Quantitative Performance Characteristics for Azodicarboxamide FTIR Analysis
| Parameter | Result | Specification |
|---|---|---|
| Linear Range | 10-40 mg/mL | - |
| Correlation Coefficient (r) | 0.9998 | n = 15 |
| Detection Limit for Impurities | 5.0 μg/g | - |
| Quantitation Limit for Impurities | 15 μg/g | - |
| Characteristic Spectral Region | 2000-1500 cm⁻¹ | DMSO solvent compatibility |
Results and Interpretation
The method demonstrated excellent linearity across the specified concentration range with a correlation coefficient of 0.9998, indicating strong predictive accuracy for API quantification [55]. The selected spectral region (2000-1500 cm⁻¹) provided optimal discrimination between API and common excipients, with no interference from flowing agents in dosage forms. Impurity profiling in non-polar solvents enabled detection down to 5.0 μg/g, sufficient for monitoring potential synthesis by-products.
The experimental workflow for this comprehensive analysis is systematized below:
Advanced FTIR Applications in Formulation Development
Polymorph Characterization and Monitoring
Different polymorphic forms of pharmaceutical compounds significantly impact stability, bioavailability, and efficacy [52]. FTIR spectroscopy provides sensitive detection of polymorphic conversions through subtle shifts in characteristic absorption bands.
Experimental Protocol for Polymorph Screening:
- Equipment Setup: Utilize high-temperature ATR accessory capable of ramping to 300°C
- Temperature Programming: Implement controlled temperature gradients (e.g., 2-5°C/min)
- Spectral Monitoring: Collect spectra at regular temperature intervals
- Data Analysis: Identify polymorphic transitions through band shifts and appearance/disappearance of characteristic peaks
Case Example: Variable temperature ATR-FTIR using the Golden Gate High Temperature ATR Accessory successfully profiled paracetamol polymorphs, clearly identifying phase changes that challenge detection by other analytical techniques due to similar transition temperatures [52].
Drug-Excipient Compatibility Studies
FTIR spectroscopy effectively identifies molecular interactions between APIs and excipients through shifts in key spectral bands, preventing formulation failures [52] [53].
Experimental Protocol:
- Binary Mixture Preparation: Combine API with individual excipients at relevant ratios
- Stress Conditions: Expose mixtures to accelerated stability conditions (elevated temperature/humidity)
- Spectral Comparison: Analyze fresh and stressed mixtures for band shifts, disappearance, or new peaks
- Interpretation: Correlate spectral changes with potential molecular interactions
Case Example: ATR-FTIR revealed incompatibility between levodopa (Parkinson's medication) and many common excipients, guiding rational formulation development [52].
Quality Control and PAT Applications
FTIR spectroscopy supports real-time quality monitoring in pharmaceutical manufacturing through integration into PAT frameworks [52] [53].
Table 2: FTIR Quality Control Applications in Pharmaceutical Manufacturing
| Application Area | FTIR Technique | Key Performance Metrics |
|---|---|---|
| Blend Uniformity | NIR Spectroscopy | Real-time homogeneity assessment |
| Moisture Analysis | DRIFTS | 2-20% moisture range in tablets |
| API Identity Verification | ATR-FTIR | Rapid spectral fingerprint matching |
| Counterfeit Detection | ATR-FTIR | Compositional differences in 1800-525 cm⁻¹ region |
| Foreign Particulate Analysis | FTIR Microscopy | Identification of proteinaceous/non-proteinaceous particles |
Case Example: DRIFTS provided a non-destructive alternative to Karl Fischer titration for moisture analysis in 5-fluorouracil chemotherapy drug, enabling rapid quality assessment without sample destruction [52].
NMR Spectroscopy in Functional Group Analysis
Complementary Role in Structural Elucidation
While FTIR provides information about functional groups based on bond vibrations, NMR spectroscopy offers atomic-level structural insights through detection of magnetic properties of specific nuclei (¹H, ¹³C, ¹⁵N, etc.). The combination of these techniques delivers comprehensive molecular characterization unattainable by either method alone [5].
Experimental Considerations for Pharmaceutical Analysis
Sample Preparation:
- Solvent Selection: Use deuterated solvents matching reference spectra conditions (e.g., CDCl₃)
- Concentration Optimization: Balance between signal-to-noise and saturation effects
- Temperature Control: Maintain consistent temperature for chemical shift reproducibility
- Reference Standards: Include internal standards (TMS for ¹H/¹³C NMR) for chemical shift calibration
Data Acquisition Parameters:
- Spectral Width: Appropriate to expected chemical shift range
- Acquisition Time: Sufficient for adequate digital resolution
- Relaxation Delays: Allow complete spin-lattice relaxation for quantitative accuracy
- Number of Scans: Optimize based on sample concentration and instrument sensitivity
Integrated FTIR-NMR Approach with Machine Learning
Enhanced Functional Group Identification
Recent advances in spectroscopic analysis incorporate machine learning to improve accuracy and efficiency of functional group identification from FTIR and NMR data [5]. Artificial neural network (ANN) models trained on multiple spectroscopic data types demonstrate superior performance compared to single-technique approaches.
Experimental Protocol for Integrated Analysis:
Data Collection:
- FTIR spectra (400-4000 cm⁻¹ range, 3.25 cm⁻¹ resolution)
- ¹H NMR spectra (1-12 ppm range, 1 ppm binning intervals)
- ¹³C NMR spectra (1-220 ppm range, 5 ppm binning intervals)
Data Preprocessing:
- FTIR: Convert transmittance to absorbance, apply min-max normalization
- NMR: Apply data binning, peak presence/absence encoding (1/0)
- Handle missing values through linear interpolation
Model Training:
- Implement artificial neural network with input layers for each spectral type
- Apply stratified 5-fold cross-validation to prevent overfitting
- Optimize weights and thresholds to minimize prediction errors
Table 3: Performance Comparison of Functional Group Identification Methods
| Analytical Approach | Macro-average F1 Score | Key Advantages |
|---|---|---|
| FTIR Spectroscopy Only | 0.88 | Rapid analysis, minimal sample preparation |
| NMR Spectroscopy Only | 0.85 | Detailed structural information |
| Combined FTIR-NMR with ANN | 0.93 | Superior accuracy, comprehensive molecular characterization |
Case Study: Machine Learning-Enabled Functional Group Prediction
A 2025 study demonstrated that ANN models trained on combined FTIR-NMR spectral data identified 17 functional groups with significantly improved accuracy compared to single-technique models [5]. The integrated approach particularly enhanced detection of challenging functional groups including nitriles, alkyl halides, and ethers that often exhibit weak spectroscopic signals.
The workflow for this integrated machine learning approach is visualized below:
Emerging Applications and Future Directions
Point-of-Care Analysis of 3D Printed Dosage Forms
As personalized medicine advances, FTIR spectroscopy shows promise for quality control of 3D printed dosage forms in point-of-care clinical settings [52] [53]. Preliminary research with griseofulvin, indomethacin, and nifedipine formulations demonstrates feasibility for rapid verification of API identity and distribution in customized dosage forms.
Biologics and RNA Therapeutics Characterization
FTIR spectroscopy increasingly supports biologics formulation development through analysis of secondary structure in protein-based therapeutics [52] [56]. The technique also shows potential for RNA therapeutics characterization, with demonstrated sensitivity to RNA structure in basic research applications [52].
High-Resolution FTIR Microscopy
Advanced FTIR microscopy systems like the Nicolet RaptIR FTIR Microscope enable simultaneous macro and micro analysis of pharmaceutical samples [56]. Applications include:
- Foreign particulate identification in parenteral formulations
- Drug distribution mapping in inhaled products
- Agglomeration characterization in suspension formulations
- Chemical imaging of heterogeneous dosage forms
Essential Research Reagent Solutions
Table 4: Key Materials and Reagents for Pharmaceutical FTIR/NMR Analysis
| Item | Function | Application Notes |
|---|---|---|
| DMSO-d⁶ | NMR Solvent | Ideal for polar compounds, provides lock signal |
| CDCl₃ | NMR Solvent | Standard for organic molecules, referenced in databases |
| CaF₂ Windows | Liquid Cell Material | Transparent to IR, suitable for aqueous samples |
| Diamond ATR Crystal | Solid Sample Analysis | Durable, chemically resistant, minimal sample prep |
| ZnSe Crystal Rod | CIR Accessory | Provides precise 13 μm pathlength for quantitative work |
| Teflon Spacers | Liquid Cell Assembly | Define pathlength, compatible with most solvents |
| Tetramethylsilane (TMS) | NMR Reference Standard | 0 ppm chemical shift reference for ¹H and ¹³C NMR |
| NIST Traceable Standards | Instrument Calibration | Verify wavelength accuracy and photometric linearity |
The integrated application of FTIR and NMR spectroscopy provides a powerful analytical framework for comprehensive functional group analysis throughout pharmaceutical development. From initial API characterization to final product quality control, these techniques deliver complementary molecular insights that support formulation optimization, stability assessment, and regulatory compliance. The emerging integration of machine learning approaches with multimodal spectroscopic data further enhances analytical capabilities, enabling more accurate and efficient structural elucidation of complex pharmaceutical compounds. As the industry advances toward continuous manufacturing and personalized medicine, FTIR and NMR spectroscopy will continue to play critical roles in ensuring drug safety, efficacy, and quality.
Hyphenated techniques represent a powerful paradigm in modern analytical chemistry, developed from the coupling of a separation technique with an on-line spectroscopic detection technology [57]. The term "hyphenation" was introduced to refer to the on-line combination of a separation technique like gas chromatography (GC) or liquid chromatography (LC) with one or more spectroscopic detection techniques such as infrared (IR) or nuclear magnetic resonance (NMR) spectroscopy [57]. These techniques exploit the advantages of both methodologies: chromatography produces pure or nearly pure fractions of chemical components in a mixture, while spectroscopy provides selective information for identification using standards or library spectra [57].
The remarkable improvements in hyphenated analytical methods over recent decades have significantly broadened their applications across numerous fields, including the analysis of biomaterials, natural products, pharmaceuticals, polymers, and environmental samples [57] [58]. The power of combining separation technologies with spectroscopic techniques has been demonstrated for both quantitative and qualitative analysis of unknown compounds in complex mixtures such as natural product extracts or industrial polymer formulations [57] [58]. In the specific context of functional group research, these techniques provide unparalleled capability for identifying and characterizing the specific atomic arrangements that dictate molecular properties and reactivity.
Gas Chromatography-Infrared Spectroscopy (GC-IR)
Principles and Instrumentation
GC-IR combines the high-efficiency separation capability of gas chromatography with the molecular structure identification power of infrared spectroscopy [59]. In this technique, the gas chromatograph serves as a pre-separation tool for the infrared spectrometer, while the infrared spectrometer acts as a specialized qualitative detector for the chromatographic separation [59]. The development of Fourier Transform Infrared (FTIR) spectroscopy was pivotal for enabling GC-IR, as FTIR provides the fast scanning speed and high sensitivity necessary for real-time monitoring of chromatographic fractions [59].
A typical GC-FTIR system consists of four main components: the gas chromatograph for separating mixture components; the interface (most commonly a light pipe interface); the Fourier transform infrared spectrometer; and the computer data system for controlling online operation and processing data [59]. The working principle involves the sample being separated by GC, after which each fraction enters the interface sequentially according to retention time. The fractions selectively absorb infrared light in the interface, generating interference signals detected by the infrared detector. The computer system stores the collected interferogram information and converts it into gaseous infrared spectra through fast Fourier transformation [59].
Applications and Protocols
Protocol for Natural Product Volatile Oil Analysis [59]:
- Sample Preparation: Dissolve volatile oil samples in appropriate organic solvents (e.g., dichloromethane or hexane) at concentrations typically ranging from 1-10 mg/mL.
- GC Conditions: Use capillary GC columns (e.g., 30m × 0.25mm ID) with stationary phases appropriate for the analyte polarity. Employ temperature programming from 40°C (hold 2 min) to 280°C at 5-10°C/min. Use helium carrier gas with split or splitless injection modes.
- Interface Settings: Maintain light pipe temperature at 250-300°C to prevent analyte condensation.
- FTIR Parameters: Set resolution to 4-8 cm⁻¹ with scan rates of 1-2 spectra per second to adequately track eluting peaks.
- Data Analysis: Compare obtained gaseous infrared spectra with standard library spectra (e.g., EPA vapor phase database) for compound identification.
GC-IR finds particular utility in distinguishing between isomeric compounds that may have similar chromatographic behavior but distinct infrared spectral signatures [59]. The technique provides several types of information for each component: chromatographic retention values, reconstructed chromatograms, infrared spectra, and library search results with match scores [59].
Table 1: Key Applications of GC-IR in Functional Group Analysis
| Application Area | Specific Uses | Functional Groups Identified |
|---|---|---|
| Natural Product Volatile Oils | Medicinal volatile oil analysis, quality control | Hydroxyl groups (alcohols, phenols), carbonyls (aldehydes, ketones), ether linkages |
| Flavors and Fragrances | Isomer separation, complex mixture analysis | Esters, aldehydes, ketones, alcohols, terpene functionalities |
| Environmental Analysis | Pollutant identification, pesticide residue detection | Chlorinated hydrocarbons, phosphates, carbamates |
| Petrochemical | Hydrocarbon analysis, oxygenate identification | Alkanes, alkenes, aromatics, alcohols, ethers |
Research Reagent Solutions
Table 2: Essential Research Reagents for GC-IR Analysis
| Reagent/Material | Function | Application Notes |
|---|---|---|
| High-purity helium carrier gas | Mobile phase for GC separation | Must be oxygen-free to protect columns and prevent degradation |
| Deuterated solvents (e.g., CDCl₃) | Sample preparation for NMR-correlative studies | For complementary structural analysis |
| Trimethylsilyl derivatization agents | Enhance volatility of polar compounds | For analysis of hydroxyl-containing compounds (alcohols, phenols, carboxylic acids) |
| Capillary GC columns (various phases) | Compound separation based on polarity/volatility | Selection depends on analyte characteristics |
| FTIR calibration standards (e.g., polystyrene) | Instrument performance verification | Ensure spectral accuracy and reproducibility |
Thermogravimetric Analysis-Infrared Spectroscopy (TGA-IR)
Principles and Instrumentation
TGA-IR combines thermal gravimetric analysis, which measures small weight losses during a controlled heating ramp, with Fourier transform infrared spectroscopy for identification of the evolved gases [60]. While TGA provides quantitative information about weight loss at specific temperatures, it cannot identify the chemicals responsible for these weight losses [60]. The integration of FTIR as a detector solves this limitation by providing chemical identification capabilities for the evolved gases.
The instrumentation consists of a TGA unit coupled to an FTIR spectrometer via a heated transfer line that connects the TGA outlet to an IR gas cell [60]. The transfer line is typically maintained at temperatures up to 300°C to prevent condensation of the evolved gases [60]. During analysis, the FTIR collects spectra continuously (typically every 5-10 seconds) throughout the TGA temperature ramp, generating a three-dimensional data set of frequency versus intensity versus time [60].
Applications and Protocols
Protocol for Polymer Deformulation [60]:
- Sample Preparation: Place 10-20 mg of polymer sample into TGA platinum crucible. Ensure homogeneous sample distribution for consistent thermal degradation.
- TGA Parameters: Set temperature ramp from 35°C to 800°C at 10-20°C/min under nitrogen or air atmosphere (flow rate: 50-100 mL/min). Select atmosphere based on desired degradation pathway (inert for pyrolysis, oxidative for combustion studies).
- Transfer Line Conditions: Maintain temperature at 250-300°C to prevent analyte condensation.
- FTIR Settings: Equip with liquid nitrogen-cooled MCT detector for high sensitivity. Set resolution to 4 cm⁻¹ with 8-16 scans co-added per spectrum.
- Data Analysis: Use multivariate curve resolution (MCR) algorithms to deconvolute overlapping evolution profiles and identify individual components through spectral library matching.
The TGA-IR data analysis challenge is substantial, as a typical experiment lasting 30-60 minutes can generate 500-800 spectra representing complex mixtures of evolved gases [60]. Advanced data processing techniques like the Mercury TGA algorithm automate the analysis by performing multivariate curve resolution to extract linearly independent spectral features called "Pure Component" spectra [60]. This approach creates time-evolving profiles for each identified component, revealing when a gas starts evolving and the duration of its evolution [60].
Diagram 1: TGA-IR Workflow for Functional Group Analysis
Characteristic Functional Group Identifiers
Table 3: Characteristic IR Bands for Functional Group Identification in TGA-IR
| Functional Group | Characteristic IR Bands (cm⁻¹) | Associated Materials/Compounds |
|---|---|---|
| Hydroxyl (-OH) | 3200-3650 (broad) | Alcohols, water, carbohydrates |
| Carbonyl (C=O) | 1650-1800 | Esters, ketones, carboxylic acids, anhydrides |
| Carbon dioxide | 2349, 667 | Carbonate decomposition, combustion product |
| Ester (C-O-C) | 1050-1300 | Polyesters, plasticizers |
| Amine (N-H) | 3300-3500 | Polyamides, proteins |
| Ether (C-O-C) | 1070-1150 | Polyethers, epoxy resins |
| Nitrile (C≡N) | 2200-2260 | Acrylonitrile polymers |
| Aromatic (C-H) | 3000-3100, 1600, 1500 | Polystyrene, phenolic resins |
Liquid Chromatography-Nuclear Magnetic Resonance (LC-NMR)
Principles and Instrumentation
LC-NMR combines the outstanding separation power of liquid chromatography with the superior structural elucidation capability of nuclear magnetic resonance spectroscopy [61]. This hyphenated technique has evolved from an academic curiosity to a robust analytical tool, particularly valuable for natural product analysis and pharmaceutical applications [61]. NMR stands out as a detector for LC by providing maximum structural information about plant-originated extracts, particularly regarding the isolating ability of isomeric and/or isobaric compounds compared to other detection methods [61].
The general instrumentation of an LC-NMR system includes three main zones: the isolation zone (chromatography column), the interface zone, and the detection zone (NMR probe) [61]. The HPLC system is directly connected to the NMR under computer-controlled data acquisition with automated harmonization of different operations [61]. Sensitive detectors such as UV and/or MS are often coupled in parallel with proper splitting ratios to guide NMR measurements [61].
Operational Modes
LC-NMR can operate in several modes, each with specific advantages for functional group research:
On-flow Mode (Continuous Flow): The simplest setup where spectra are acquired continuously as compounds elute from the LC column. This mode maintains good separation resolution but suffers from relatively poor sensitivity due to short detection times and potential solvent signal interference [61].
Stop-flow Mode: The chromatographic flow is stopped when a peak of interest reaches the NMR flow cell, allowing extended acquisition times for improved signal-to-noise ratio. This approach enables more sophisticated 2D NMR experiments but requires adequate separation between peaks [61].
Loop-storage/Cartridge Mode (LC-SPE-NMR): A advanced approach where eluted peaks are stored in loops or solid-phase extraction (SPE) cartridges after chromatographic separation. After completion of the separation, the stored peaks are transferred to the NMR flow cell using deuterated solvents. This approach significantly reduces solvent costs and enhances sensitivity through sample concentration [61].
Diagram 2: LC-NMR Operational Modes for Functional Group Characterization
Applications and Protocols
Protocol for Natural Product Analysis Using LC-SPE-NMR [61]:
- Sample Preparation: Extract natural product material (plant, microbial) with appropriate solvents (methanol, ethanol, or hydroalcoholic mixtures). Concentrate under reduced temperature and pressure.
- LC Conditions: Use reversed-phase columns (C18, 150-250 mm × 2-4.6 mm) with water-acetonitrile or water-methanol gradients. Employ trifluoroacetic acid or formic acid (0.1%) as mobile phase modifier to enhance separation.
- SPE Cartridge Conditioning: Condition SPE cartridges (typically C18 or polymeric sorbents) with deuterated methanol followed by deuterium oxide.
- Peak Trapping: Direct LC eluent to divert to waste or SPE cartridges based on UV or MS triggering. Capture multiple injections to enhance analyte concentration.
- Cartridge Drying: Remove residual protonated solvents by passing dry nitrogen gas through cartridges for 20-30 minutes.
- NMR Analysis: Elute trapped analytes directly into NMR flow cell using deuterated solvents (CD3OD, CDCl3, or DMSO-d6). Acquire 1D and 2D NMR experiments (1H, 13C, COSY, HSQC, HMBC) for complete structural elucidation.
LC-NMR has demonstrated particular utility in identifying isoflavonoids from plant sources such as Smirnowia iranica, where it enabled the characterization of ten new compounds in addition to seven previously known constituents [61]. The technique provides detailed information about functional groups including hydroxyl, carbonyl, aromatic, and glycosidic linkages through chemical shift analysis and through-bond correlations in 2D experiments.
Table 4: NMR Chemical Shifts for Key Functional Groups in Natural Product Analysis
| Functional Group | ¹H Chemical Shift (δ, ppm) | ¹³C Chemical Shift (δ, ppm) | Structural Significance |
|---|---|---|---|
| Aliphatic alcohol (-OH) | 1.0-5.5 (variable) | 50-90 (C-OH) | Carbohydrates, glycosides |
| Phenolic hydroxyl (-OH) | 4.5-9.0 (variable) | 140-165 (C-OH) | Flavonoids, tannins |
| Aldehyde (-CHO) | 9.0-10.0 | 185-205 | Terpenoids, flavor compounds |
| Carboxylic acid (-COOH) | 10.0-13.0 | 170-185 | Organic acids, fatty acids |
| Aromatic | 6.0-8.5 | 110-160 | Phenolic compounds, alkaloids |
| Methoxy (-OCH₃) | 3.3-4.0 | 50-60 | Lignans, flavonoids |
| Methylene (-CH₂-) | 1.2-1.4 | 20-50 | Aliphatic chains |
| Carbonyl (amide) | 5.5-8.5 (NH) | 165-180 | Peptides, proteins |
Comparative Analysis and Emerging Trends
Technique Selection Guidelines
Each hyphenated technique offers distinct advantages for functional group analysis. GC-IR provides excellent sensitivity for volatile compounds and unambiguous identification of isomers through gas-phase IR spectra [59]. TGA-IR uniquely couples thermal properties with chemical identification, making it invaluable for studying decomposition processes and polymer stability [60]. LC-NMR delivers the most comprehensive structural information, particularly for isomeric and isobaric compounds that challenge mass spectrometric techniques [61].
Recent technological advancements have significantly enhanced the capabilities of these hyphenated systems. In LC-NMR, the development of cryogenic probe and microprobe technologies has dramatically improved sensitivity, enabling the analysis of smaller sample quantities [61]. For GC-IR and TGA-IR, the integration of multivariate curve resolution algorithms has transformed data analysis, allowing deconvolution of complex overlapping signals [60].
Integration with Machine Learning Approaches
Emerging approaches in functional group identification involve machine learning models that simultaneously train on multiple spectroscopic data types [5]. Recent research demonstrates that artificial neural network models trained on combined FT-IR, ¹H NMR, and ¹³C NMR data can identify 17 functional groups with a macro-average F1 score of 0.93, outperforming models using single spectroscopic techniques [5]. This integrated analytical approach mirrors how expert spectroscopists naturally combine information from multiple techniques for more accurate structural analysis.
The future development of hyphenated techniques will likely focus on increasing sensitivity, reducing solvent consumption, and enhancing data integration capabilities. Techniques such as capLC-NMR and comprehensive LC×LC-NMR represent promising directions for analyzing limited sample quantities [61]. Additionally, the integration of machine learning algorithms for real-time spectral interpretation will further streamline the identification of functional groups in complex mixtures [5].
Application Note: FT-IR Spectroscopy for Protein Dynamics and H/D Exchange
Protein function is intrinsically linked to spatial and temporal structural changes, collectively known as protein dynamics. Fourier-Transform Infrared (FT-IR) spectroscopy serves as a powerful tool for probing these dynamics, particularly through amide hydrogen/deuterium (H/D) exchange experiments. This protocol outlines the steps for utilizing FT-IR spectroscopy to monitor protein dynamics, applicable for studying the effects of mutations, ligand interactions, or metal ion binding on protein structural stability and flexibility [62].
Detailed Experimental Protocol
Protein Sample Preparation
- Sample Purity: Begin with a purified protein solution. Ensure the buffer is compatible with FT-IR analysis, typically using low-concentration salts like phosphate or Tris to minimize infrared absorption interference.
- Deuterated Buffer Preparation: Prepare a deuterated buffer (e.g., D₂O-based phosphate buffer) for the H/D exchange process. Pre-equilibrate the pH meter reading for D₂O (pD ≈ pH reading + 0.4).
- H/D Exchange Initiation: Rapidly mix the protein solution with the deuterated buffer. The typical protein concentration for FT-IR analysis ranges from 1 to 10 mg/mL. Immediately after mixing, load the sample into a sealed FT-IR liquid cell with appropriate pathlength (e.g., 50 µm CaF₂ windows).
FT-IR Spectra Collection
- Instrument Setup: Use an FT-IR spectrometer equipped with a liquid nitrogen-cooled MCT (Mercury Cadmium Telluride) detector for high sensitivity.
- Data Acquisition Parameters:
- Spectral Range: 1000 cm⁻¹ to 4000 cm⁻¹, with focus on the amide I (1600-1700 cm⁻¹) and amide II (1480-1580 cm⁻¹) regions.
- Resolution: Set to 4 cm⁻¹.
- Scans: Accumulate 128-256 scans per spectrum to achieve a high signal-to-noise ratio.
- Kinetic Data Collection: Collect spectra sequentially over time immediately after initiating H/D exchange. The time intervals can range from seconds to hours, depending on the exchange kinetics of the protein system.
- Control Measurements: Acquire spectra of the protein in non-deuterated buffer and the deuterated buffer alone for background subtraction.
Spectra Analysis and Data Processing
- Pre-processing: Subtract the buffer spectrum from the protein spectrum at each time point. Apply atmospheric correction (for CO₂ and water vapor) if necessary.
- Peak Intensity Monitoring: The primary data for H/D exchange kinetics is the decrease in intensity of the amide II band (∼1550 cm⁻¹), which arises mainly from N-H bending vibrations. As H is exchanged for D, this band diminishes while the amide II' band (∼1450 cm⁻¹) increases.
- Kinetic Parameter Fitting: Normalize the amide II band intensity at time t (It) to the initial intensity (I₀). Plot (It / I₀) versus time and fit the decay to an exponential model to obtain the exchange rate constant(s). For multi-phase kinetics, use a multi-exponential fit: I_t / I₀ = A₁exp(-k₁t) + A₂exp(-k₂t) + ... where Aᵢ are amplitudes and kᵢ are the rate constants for different phases of exchange.
Research Reagent Solutions
Table 1: Essential Reagents for Protein Dynamics Analysis via FT-IR
| Reagent/Material | Function | Example/Note |
|---|---|---|
| Purified Protein | Analyte of interest | ≥95% purity recommended; concentration 1-10 mg/mL |
| Deuterium Oxide (D₂O) | Deuterium source for H/D exchange | Enables tracking of backbone dynamics |
| CaF₂ or BaF₂ Windows | IR-transparent cell windows | For sample containment in liquid cell |
| Compatible Buffer Salts | Maintain protein stability and pH/pD | e.g., phosphate, Tris; in low concentration |
Application Note: FT-IR and NMR with Machine Learning for Functional Group Identification
The identification of functional groups is a fundamental task in chemical analysis, critical for elucidating the structure and properties of unknown compounds. While FT-IR and NMR spectroscopy are cornerstone techniques for this purpose, interpreting their data can be challenging and time-consuming. Recent advances have integrated machine learning with complementary spectroscopic data to achieve rapid, accurate functional group identification, outperforming traditional analysis methods [5].
Detailed Protocol for ML-Enhanced Functional Group Analysis
Data Collection and Preprocessing
- FT-IR Data Handling:
- Collect FT-IR spectra in the range of 400–4000 cm⁻¹.
- Convert transmittance values to absorbance.
- Transform each spectrum into a vector of 1108 data points representing wavelengths from 400 to 4000 cm⁻¹ with a 3.25 cm⁻¹ resolution.
- Apply min-max normalization by dividing each absorbance value by the maximum absorbance value in the dataset.
- NMR Data Handling (¹H and ¹³C):
- For ¹H NMR, use the spectral range of 1–12 ppm. Divide into 12 bins, each of 1 ppm interval.
- For ¹³C NMR, use the spectral range of 1–220 ppm. Divide into 44 bins, each of 5 ppm interval.
- Assign a value of 1 if a peak is present in a particular bin, and 0 if absent (intensity values are ignored to mitigate data sparsity issues).
- Data Integration: Concatenate the processed FT-IR and NMR vectors to form a combined input for the machine learning model.
Machine Learning Model Training
- Model Architecture: Employ an Artificial Neural Network (ANN). The input layer size corresponds to the total dimensions of the combined spectral data (1108 from FT-IR + 12 from ¹H NMR + 44 from ¹³C NMR = 1164 nodes).
- Training Configuration:
- Use multiple hidden layers with activation functions (e.g., ReLU).
- Apply batch normalization in each layer to stabilize training.
- Implement a stratified 5-fold cross-validation on 80% of the data to prevent overfitting, using the remaining 20% as a test set.
- Output Layer: Configure the output layer with 17 nodes, each corresponding to one of the following functional groups: aromatic, acyl halide, ether, alcohol, ester, methyl, nitro, alkane, carboxylic acid, amine, aldehyde, alkyne, ketone, alkyl halide, amide, alkene, and nitrile. Use a sigmoid activation function for multi-label classification.
Functional Group Prediction and Validation
- Prediction: Input the preprocessed, combined spectral data of an unknown compound into the trained ANN model.
- Output Interpretation: The model outputs a probability (between 0 and 1) for the presence of each of the 17 functional groups. A threshold (e.g., 0.5) is applied to make a final classification.
- Validation: The model's performance is evaluated using the macro-average F1 score on the test set. The integrated FT-IR/NMR model has been shown to achieve an F1 score of 0.93, significantly outperforming models using FT-IR data alone (F1 score 0.88) [5].
Workflow Diagram: ML-Driven Functional Group Analysis
Application Note: FT-IR Spectroscopy for Microplastic Analysis in Drinking Water
Microplastics (MPs), defined as plastic particles smaller than 5 mm, are pervasive environmental pollutants detected in various water sources, including those intended for human consumption. FT-IR spectroscopy is a gold-standard technique for the identification and quantification of microplastics due to its ability to provide a unique molecular fingerprint of polymer types. Standardizing methodologies is crucial for generating comparable data and assessing human exposure risks [63] [64].
Detailed Protocol for Microplastic Detection
Sample Collection and Filtration
- Water Sampling: Collect drinking water samples (e.g., 1 L volume) in clean glass containers to avoid contamination.
- Filtration Setup: Based on meta-analysis findings, use a cellulose nitrate membrane filter with a pore size of 0.45 µm, placed in a Büchner funnel connected to a vacuum filtration system [64].
- Filtration Process: Filter the water sample through the membrane. The cellulose nitrate filter is recommended for its superior retention capabilities for microplastics.
Sample Staining and Preparation
- Nile Red Staining: To facilitate fluorescent detection and counting, add Nile red dye (1 mg/L in methanol) to the water sample before or after filtration. For post-filtration staining, the filter with the retained particles can be stained.
- Incubation: Place the filter in an oven at 30°C for 30 minutes to allow the dye to bind to the polymers, inducing fluorescence.
- Blank Controls: Process blank samples containing 250 mL of distilled water with Nile red dye and methanol through the same filtration and staining procedure to account for potential background contamination.
FT-IR Analysis and Identification
- Instrumentation: Use an FT-IR spectrometer equipped with a microscope for microspectroscopic analysis (µFT-IR). Transmission or reflectance modes can be used.
- Spectral Acquisition:
- Spectral Range: 4000–600 cm⁻¹.
- Resolution: 4–8 cm⁻¹.
- Scans: 32–64 scans per spectrum.
- Particle Analysis: Manually or automatically locate particles on the filter. Acquire IR spectra of individual particles.
- Spectral Interpretation: Compare the acquired spectra against reference spectral libraries of known polymers (e.g., polyethylene (PE), polypropylene (PP), polystyrene (PS), polyvinyl chloride (PVC)). A successful match confirms the polymer identity of the microplastic.
Quantitative Data on Method Comparison
Table 2: Key Techniques for Microplastic Analysis
| Technique | Key Advantage | Key Limitation | Typical Use Case |
|---|---|---|---|
| FT-IR Spectroscopy | Broad application range; polymer identification; can determine size/shape [63] | Limited for very small MPs (<20 µm); can struggle with mixtures [63] | Standard analysis for MPs in water; polymer sourcing [64] |
| Raman Spectroscopy | Excels at detecting smaller particles; high spatial resolution [63] | Can suffer from fluorescence interference [63] | Complementary to FT-IR for sub-micron particles |
| Pyrolysis-GC–MS | Chemical identification; good for complex mixtures | Destructive technique; requires expertise | Detailed chemical analysis of polymer composition |
Research Reagent Solutions
Table 3: Essential Reagents for Microplastic Analysis via FT-IR
| Reagent/Material | Function | Example/Note |
|---|---|---|
| Cellulose Nitrate Filter | Retains microplastics from liquid sample | 0.45 µm pore size is optimal [64] |
| Nile Red Dye | Fluorescent staining of plastic particles | Aids visual detection and counting [64] |
| Methanol | Solvent for Nile Red dye | Laboratory grade |
| Reference Polymer Libraries | Spectral database for polymer identification | Essential for accurate material ID |
Emerging Frontier: Clinical Diagnostics and Toxicological Assessment
In silico Analysis of Microplastic-Protein Interactions
The concern regarding microplastics extends beyond environmental presence to their potential impacts on human health. Computational studies are emerging to understand the molecular basis of these interactions. Molecular docking analyses have been employed to investigate the binding of common microplastics (e.g., polystyrene, polycarbonate, polyethylene terephthalate) to human proteins, such as Cytochrome P450 1A1 (CYP1A1), a key enzyme in xenobiotic detoxification [65] [66].
- Findings: Studies indicate that certain MPs, like polycarbonate (binding affinity ΔG = -7.4 kcal/mol) and polyethylene terephthalate (ΔG = -7.1 kcal/mol), can bind to the active site of CYP1A1 with significant affinity, primarily via hydrophobic interactions and hydrogen bonding [66].
- Implication: This suggests a potential for MPs to disrupt normal metabolic processes, alter the enzyme's activity towards other toxins, and contribute to toxicological effects. This computational approach provides a mechanistic framework for guiding future in vitro and in vivo toxicological studies [65].
Workflow Diagram: Microplastic Analysis & Risk Assessment
Enhancing Accuracy: Overcoming Common Challenges and Data Pitfalls
Resolving Spectral Ambiguities and Weak Signals in FTIR and NMR
In the fields of analytical chemistry and drug development, Fourier Transform Infrared (FTIR) spectroscopy and Nuclear Magnetic Resonance (NMR) spectroscopy are indispensable tools for qualitative functional group analysis and molecular structure elucidation. However, a significant and common challenge faced by researchers is the reliable interpretation of data comprised of weak signals and ambiguous spectral features. These issues are particularly prevalent when analyzing complex mixtures, samples at low concentrations, or molecules in dynamic states, which can obscure the definitive identification of functional groups. In FTIR, spectral complexities often arise from overlapping vibrational bands in multi-component systems, while in NMR, low sensitivity and signal-to-noise ratio can hinder the detection of key nuclei. This application note details advanced, practical methodologies to overcome these hurdles, leveraging multivariate analysis for FTIR and novel resonance techniques for NMR, thereby enabling researchers to extract more meaningful information from their spectroscopic data.
Overcoming FTIR Spectral Ambiguities in Complex Mixtures
The analysis of complex mixtures using FTIR spectroscopy often results in heavily overlapped spectral bands, making it difficult to resolve the individual components. This is a classic example of spectral ambiguity.
Multivariate Curve Resolution – Alternating Least Squares (MCR-ALS)
Principle: MCR-ALS is a powerful chemometric technique that decomposes a set of spectra from a mixture system into the pure spectral profiles and their corresponding concentration profiles without prior knowledge of the system's composition [67]. It is particularly effective for extracting information from evolving systems, such as reaction monitoring or the study of adulterated products.
A recent study on detecting adulterants in virgin coconut oil (VCO) provides an excellent protocol for implementing MCR-ALS [67]. The economic incentive to adulterate VCO with cheaper oils like sunflower (SO), maize (MO), and peanut (PO) oil necessitates robust analytical techniques. The substantial spectral overlap between these oils makes traditional univariate analysis unsuitable.
Table 1: MCR-ALS Protocol for Resolving FTIR Spectra of Oil Adulterants
| Step | Action | Details and Parameters |
|---|---|---|
| 1. Data Collection | Acquire FTIR spectra of pure and adulterated samples. | Use an ATR accessory for rapid analysis; spectral range: 3100-2500 cm⁻¹ and 1900-450 cm⁻¹ [67]. |
| 2. Data Pre-processing | Apply derivatization and standard normal variate (SNV). | Use the Savitzky-Golay algorithm for derivatization to resolve overlapping peaks and enhance spectral features [67]. |
| 3. Data Arrangement | Construct a column-wise augmented matrix. | Matrix includes a training set (pure VCO and known adulterated samples) and a validation set (unknowns) for classification [67]. |
| 4. MCR-ALS Modeling | Decompose the data matrix with constraints. | Apply non-negativity constraints to both concentration and spectral profiles during the Alternating Least Squares optimization [67]. |
| 5. Model Optimization | Implement variable selection. | Use a Genetic Algorithm (GA) to select the most informative spectral variables, improving model parsimony and predictive ability [67]. |
| 6. Validation & Quantification | Assess model performance. | Use metrics like RMSEP (Root Mean Square Error of Prediction); reported values were 1.79-2.66 for adulterant quantification [67]. |
The MCR-ALS scores can be used to create a control chart for distinguishing pure VCO from adulterated samples with 100% correct classification in external validation [67]. Furthermore, the correlation-constrained MCR-ALS models successfully quantified the blend composition of adulterants with absolute errors of less than 4.6%.
Supplementary Techniques for FTIR Mixture Analysis
Beyond MCR-ALS, several established techniques can be employed to tackle mixture analysis, especially when full multivariate modeling is not immediately feasible.
Table 2: Five Practical Techniques for FTIR Mixture Analysis [68]
| Technique | Description | Application Example |
|---|---|---|
| 1. Purification | Physical or chromatographic separation of mixture components before analysis. | Using GC-IR or LC-IR to separate and collect pure components for individual spectral acquisition [68]. |
| 2. Spectral Subtraction | Software-based subtraction of a known component's spectrum from the mixture spectrum. | Isolating the spectrum of an amino acid (e.g., glutamine) by subtracting the spectrum of its solvent (water) [68]. |
| 3. Library Searching | Comparing the unknown mixture spectrum against a library of pure compound spectra. | Can identify a dominant component; "subtract and search again" iteratively to identify multiple components [68]. |
| 4. Mixture Analysis Software | Proprietary software that generates and compares calculated mixture spectra from library components. | Provides a list of likely components and their approximate proportions in the mixture [68]. |
| 5. Process of Elimination | A systematic, manual approach to interpretation. | Step 1: Identify and ignore atmospheric artifacts (e.g., CO₂, H₂O). Step 2: Identify peaks from known components (e.g., solvent). Step 3: Assign remaining peaks to functional groups to deduce unknown structures [68]. |
Enhancing Weak Signal Detection in NMR Spectroscopy
The inherent low sensitivity of NMR, particularly for nuclei like ¹²⁹Xe or low-concentration analytes, often results in weak signals that are challenging to detect and interpret. Recent research has opened new pathways to overcome this limitation.
Exploiting Nonsecular Resonances
Principle: Traditional NMR relies on secular resonance, where the radio-frequency (RF) irradiation matches the Larmor frequency of the nucleus. A groundbreaking study has demonstrated that a new type of resonance, known as nonsecular resonance, can occur far from the Larmor frequency and can be used to manipulate nuclear magnetization more effectively [69]. This effect arises from normally hidden nonsecular components of the magnetic dipole-dipole coupling between spins.
Table 3: Experimental Protocol for Observing Nonsecular Resonance in ¹⁹F of CaF₂
| Step | Action | Purpose & Notes |
|---|---|---|
| 1. Polarization | Apply a high magnetic field (B₀). | Allows sufficient thermal polarization of the ¹⁹F spins to build up, creating a detectable signal [69]. |
| 2. Field Jump | Jump to a lower magnetic field. | The new field is chosen so that the Larmor frequency is about half the fixed irradiation frequency, establishing the nonsecular resonance condition [69]. |
| 3. Nonsecular Irradiation | Apply RF irradiation at the fixed frequency. | The spin system is irradiated far off its new Larmor frequency. The amplitude of the RF field must be >1% of the static field to observe the effect [69]. |
| 4. Observation Jump | Jump back to the high magnetic field. | Returns the system to the standard secular resonance condition for convenient signal detection via conventional NMR [69]. |
| 5. Signal Detection | Observe the ¹⁹F NMR signal. | A strong resonant decrease in the NMR signal at the specific second field confirms the nonsecular resonance [69]. |
This method, demonstrated on ¹⁹F nuclei in fluorite (CaF₂), provides a new avenue for enhancing spin manipulation. It is anticipated that this principle could be extended to enhance the sensitivity of other nuclei, potentially by irradiating one element to affect the signal of another, and may find even stronger effects in Electron Paramagnetic Resonance (EPR) due to larger electron magnetic moments [69].
Signal Enhancement via Metal-Organic Frameworks (MOFs)
Principle: For hyperpolarized noble gases like ¹²⁹Xe, which are used as biosensors, the weak signal from xenon atoms trapped in molecular cages can be a major limitation. A novel approach involves using engineered multivariate Metal-Organic Frameworks (MOFs) to enhance the signal.
Protocol: Researchers introduced nickel into the structure of ZIF-8 (a type of MOF) to create a multivariate ZIF-8 [70]. The incorporation of nickel increases the framework's affinity for xenon atoms and prolongs their resident time within the MOF pores. This longer residency leads to a stronger accumulated NMR signal. The result was an approximately 33% stronger hyperpolarized ¹²⁹Xe NMR signal compared to pristine ZIF-8 [70]. This strategy provides a general method for enhancing sensitive ¹²⁹Xe-based molecular imaging applications in vivo.
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful implementation of the protocols described above requires specific materials and software tools.
Table 4: Key Research Reagent Solutions for Advanced Spectral Analysis
| Item / Reagent | Function / Application | Example / Note |
|---|---|---|
| Multivariate MOFs | Enhances hyperpolarized NMR signal by entrapping gas atoms more efficiently. | Nickel-doped ZIF-8 for ¹²⁹Xe signal enhancement [70]. |
| Chemometric Software | Performs multivariate analysis (MCR-ALS, PCA, PLS) on spectral data. | Required for deconvoluting overlapping FTIR signals; often includes GA for variable selection [67]. |
| Portable FT-IR Spectrometer | Enables rapid, non-invasive screening in clinical or field settings. | Used for high-throughput diagnostics from biofluid samples like bloodspots [8]. |
| ATR-FTIR Accessory | Allows direct analysis of solids and liquids with minimal sample preparation. | Essential for routine FTIR analysis of diverse sample types [8]. |
| Spectral Libraries | Database of reference spectra for compound identification via library searching. | Critical for the "Library Searching" and "Mixture Analysis" techniques [68]. |
| Model Compounds (e.g., CaF₂) | Well-characterized systems for testing and validating novel NMR methods. | Fluorite (CaF₂) was key for first observing nonsecular resonance due to strong ¹⁹F dipolar couplings [69]. |
Optimizing Instrument Parameters for Sensitivity and Resolution
Within the framework of qualitative analysis for functional group research, achieving optimal sensitivity and resolution is a cornerstone of reliable spectroscopic characterization. For researchers in drug development, this optimization is not merely an analytical exercise but a critical step in ensuring accurate structure elucidation of active pharmaceutical ingredients (APIs), impurities, and complex natural products [71]. This document provides detailed application notes and protocols for Fourier-Transform Infrared (FTIR) and Nuclear Magnetic Resonance (NMR) spectroscopy, two pivotal techniques in the modern scientist's toolkit. By systematically optimizing instrument parameters, scientists can unlock the full potential of these methods, obtaining spectra with superior clarity and fidelity to support robust research outcomes [72] [73].
NMR Spectroscopy: Protocols and Parameter Optimization
Nuclear Magnetic Resonance (NMR) spectroscopy provides unparalleled insights into molecular structure, conformation, and dynamics. Its application spans from simple functional group verification to the complete elucidation of complex unknown molecules [71].
Key Optimization Parameters in NMR
The sensitivity and resolution of an NMR experiment are governed by several critical acquisition parameters. A recent study applying Analytical Quality by Design (AQbD) principles to quantitative NMR method development provides a data-driven framework for optimization [74].
The table below summarizes the key parameters and their impact on spectral quality.
Table 1: Key NMR Parameters for Optimizing Sensitivity and Resolution
| Parameter | Impact on Sensitivity | Impact on Resolution | Optimization Guidance |
|---|---|---|---|
| Number of Scans (NS) | Directly proportional; more scans increase signal-to-noise. | Minimal direct impact. | Balance with total experiment time. 16-128 scans, target 52-96 for robustness [74]. |
| Relaxation Delay (D1) | Critical; insufficient delay leads to signal saturation. | Minimal direct impact. | Ensure ≥5*T1 for quantitative accuracy. 1-40 s, typical robust point ~6.9 s for ¹H, 26.6 s for ¹⁹F [74]. |
| Acquisition Time (AQ) | Indirect; longer AQ allows more signal averaging. | Improves digital resolution; longer AQ allows narrower peaks. | Set to capture full FID decay; 2-4 s typical. Robust points: 2.8 s (¹H), 4.9 s (¹⁹F) [74]. |
| Spectral Width (SW) | Indirect; narrower SW reduces noise per point. | Must be wide enough to avoid folding (aliasing). | Set to cover all expected signals with a small margin. |
| Pulse Width (P1) | Fundamental; incorrect angle reduces observable signal. | Incorrect setting can distort multiplet structure. | Calibrate for specific probe and solvent (e.g., 30° or 90° pulse). |
Experimental Protocol: AQbD-Compliant qNMR Method Development
The following protocol, adapted from a 2025 study on the anti-cancer drug Sorafenib, outlines a systematic, AQbD-compliant approach to developing a robust quantitative NMR method [74].
1. Define the Analytical Target Profile (ATP):
- Clearly state the method's purpose (e.g., "Quantify Sorafenib API in tablet formulations with ±2% accuracy").
- Identify Critical Method Attributes (CMAs), such as accuracy, precision, and sensitivity.
2. Risk Assessment & Parameter Screening:
- Use tools like Fishbone diagrams to identify critical analytical procedure parameters (APPs): Number of Scans, Relaxation Delay, and Acquisition Time are typically high-risk [74].
3. Experimental Design (DoE) and Modeling:
- Employ a multivariate design (e.g., Box-Behnken Design) to explore the interaction effects of the critical APPs.
- Conduct experiments as per the design and record the response (e.g., signal-to-noise ratio, peak resolution).
- Fit the data to a mathematical model and use a desirability function (e.g., Derringer's algorithm) to identify optimal parameter settings [74].
4. Define the Method Operable Design Region (MODR):
- The MODR is the multidimensional combination of APPs where the method performance criteria are met. The following workflow diagram illustrates this systematic development process.
5. Verification and Validation:
- Verify the method performance at the identified Robust Working Point (RWP) within the MODR.
- Perform full validation as per ICH Q2(R2) guidelines, including linearity, accuracy, precision, and specificity [74].
FTIR Spectroscopy: Protocols and Parameter Optimization
FTIR spectroscopy is a rapid and non-destructive technique for identifying functional groups and characterizing chemical structures based on molecular vibrations.
Key Optimization Parameters in FTIR
The primary parameters influencing FTIR spectral quality are resolution, number of scans, and the signal-to-noise ratio, which is a key performance metric of the instrument itself [73].
Table 2: Key FTIR Parameters for Optimizing Sensitivity and Resolution
| Parameter | Impact on Sensitivity | Impact on Resolution | Optimization Guidance |
|---|---|---|---|
| Resolution | Higher resolution (lower cm⁻¹) reduces energy and S/N. | Directly defines ability to distinguish close peaks. | 4 cm⁻¹ is standard for routine analysis. Use 2-8 cm⁻¹ based on sample and need [73]. |
| Number of Scans | Proportional to √N; more scans improve S/N. | No direct impact. | 16-64 scans typical. Balance time vs. S/N requirement. |
| Signal-to-Noise (S/N) Ratio | Fundamental instrument specification; higher is better. | Enables detection of weak spectral features. | Select instrument with high S/N (e.g., 55,000:1). Ensure proper maintenance [73]. |
| Apodization Function | Affects S/N and side-lobes. | Affects line shape and apparent resolution. | Norton-Beer medium is a good default. Experiment for specific needs. |
| Aperture | Larger aperture allows more light, increasing signal. | Larger aperture can degrade resolution. | Adjust based on sample and desired spot size. |
Experimental Protocol: Functional Group Analysis of an API
This protocol describes the steps for acquiring a high-quality FTIR spectrum of an active pharmaceutical ingredient for functional group identification.
1. Sample Preparation:
- KBr Pellet Method: Finely grind 1-2 mg of dry API with 100-200 mg of anhydrous potassium bromide (KBr). Press the mixture under high pressure (~8-10 tons) in a vacuum die for 1-2 minutes to form a transparent pellet.
- ATR Method: For attenuated total reflectance, simply place a small amount of the solid API directly on the ATR crystal and apply consistent pressure with the clamp.
2. Instrument Setup and Data Acquisition:
- Collect a background spectrum with the clean ATR crystal or an empty sample holder.
- Load the prepared sample.
- Set acquisition parameters:
- Spectral Range: 4000 - 400 cm⁻¹.
- Resolution: 4 cm⁻¹.
- Number of Scans: 32.
- Acquire the sample spectrum.
3. Spectral Processing and Interpretation:
- Subtract the background spectrum from the sample spectrum.
- Examine the spectrum for key functional group regions (O-H, N-H, C=O, C-O, etc.).
- Compare the spectrum to a reference standard or library spectrum for verification. The workflow for this functional group analysis is straightforward, as shown below.
The Scientist's Toolkit: Research Reagent Solutions
The following table details essential materials and reagents used in FTIR and NMR sample preparation and analysis.
Table 3: Essential Research Reagents and Materials for Spectroscopy
| Item | Function/Application | Key Consideration |
|---|---|---|
| Deuterated Solvents (DMSO-d6, CDCl3) | NMR solvent; provides a locking signal and minimizes interfering ¹H signals. | Must be of high isotopic purity (>99.8%). Hygroscopic solvents require careful handling [74]. |
| Internal Standard (e.g., Maleic Acid) | qNMR reference for quantifying the analyte of interest. | Must be chemically stable, pure, and possess a sharp, non-overlapping resonance [74]. |
| Potassium Bromide (KBr) | Matrix for preparing solid pellets for FTIR transmission analysis. | Must be anhydrous and of spectroscopic grade to avoid moisture absorption bands. |
| ATR Crystals (Diamond, ZnSe) | Enable direct solid/liquid analysis in FTIR via attenuated total reflectance. | Diamond is durable; ZnSe offers a wider spectral range but is softer and soluble in acid. |
| NMR Tubes | Holds the sample within the NMR magnet. | Quality (wall uniformity), diameter (e.g., 5 mm), and material affect spectral quality. |
In the qualitative analysis of functional groups using Fourier Transform Infrared (FT-IR) and Nuclear Magnetic Resonance (NMR) spectroscopy, sample-related challenges represent a significant hurdle to obtaining accurate and reproducible results. Solvent interference, the presence of impurities, and sample concentration effects can profoundly distort spectral data, leading to misinterpretation of molecular structure. Within the broader thesis on qualitative analysis techniques, this document establishes standardized protocols to identify, mitigate, and account for these pervasive issues. For researchers and drug development professionals, mastering these aspects is not merely procedural but fundamental to ensuring data integrity, whether for routine analysis, regulatory submissions, or groundbreaking research.
Technical Background and Data Comparison
A clear understanding of the fundamental principles and comparative strengths of FT-IR and NMR spectroscopy is essential for selecting the appropriate technique and troubleshooting sample-related issues.
Table 1: Core Principles and Sample Challenges of FT-IR and NMR Spectroscopy
| Feature | FT-IR Spectroscopy | NMR Spectroscopy |
|---|---|---|
| Basic Principle | Measures absorption of infrared light by molecular bonds, resulting in vibrational transitions [75]. | Measures the absorption of radiofrequency radiation by atomic nuclei in a magnetic field, causing spin transitions [35]. |
| Primary Output | Spectrum showing absorption intensity vs. wavenumber (cm⁻¹). | Spectrum showing resonance frequency (chemical shift, δ) vs. intensity. |
| Key Solvent Challenge | Strong absorption by solvents (especially H₂O) can obscure key regions of the sample spectrum. | Signals from protonated solvents (e.g., CHCl₃ in CDCl₃) can overlap with sample signals [76]. |
| Impurity Detection | Excellent for identifying functional groups of impurities [77]. | Highly sensitive for identifying and quantifying impurities, especially isomeric ones [71]. |
| Concentration Sensitivity | Suitable for a wide range of concentrations, including very high (~200 mg/mL) protein solutions [75]. | Requires a certain concentration for adequate signal-to-noise; high concentrations can increase viscosity and broaden signals. |
| Sample Preparation | Can use ATR mode for minimal preparation (e.g., liquids, solids). Transmission mode requires pathlength control. | Typically requires dissolution in a deuterated solvent [35]. |
Table 2: Research Reagent Solutions for Spectroscopy
| Reagent / Material | Function and Importance in Analysis |
|---|---|
| Deuterated Solvents (e.g., CDCl₃, D₂O) | Provides a deuterium lock and field frequency stabilization for NMR; minimizes intense solvent background in ¹H NMR [35] [78]. |
| Microfluidic Channels / Flow Cells | Enables in-line FT-IR monitoring of processes like chromatography; minimizes sample handling and allows for dynamic studies [75]. |
| Diamond ATR Crystal | Allows for direct measurement of solids and liquids in FT-IR with minimal sample preparation and robust performance [77]. |
| Chiral Solvating Agents | Used in NMR to elucidate stereochemistry by forming diastereomeric complexes with enantiomers, causing chemical shift differences [71]. |
| Reference Compounds (e.g., TMS) | Provides a reference peak at 0 ppm for chemical shift calibration in NMR spectroscopy [35]. |
Experimental Protocols
Protocol 1: Mitigating Solvent Interference in ATR-FT-IR Analysis of Graphene Oxide Dispersions
This protocol is adapted from machine learning-guided ATR-FT-IR studies for analyzing nanomaterials, providing a robust method to handle complex, spectrally overlapping data [77].
1. Sample Preparation:
- Prepare aqueous dispersions of the target material (e.g., graphene oxide).
- Pipette a small volume (e.g., 2-5 µL) of the dispersion directly onto the diamond crystal of an ATR accessory.
- Allow the water to evaporate at a controlled temperature (e.g., 50°C) to leave a thin film for analysis. This minimizes the strong, broad O-H absorption from water.
2. Instrumental Parameters (Bruker Vertex 70 Example):
- Spectral Range: 4000 - 400 cm⁻¹
- Resolution: 4 cm⁻¹
- Number of Scans: 64 for both sample and background
- Absorption Accessory: Single-reflection ATR with a diamond crystal
3. Data Acquisition and Machine Learning Analysis:
- Collect spectra of pristine, purified, and reduced samples.
- For complex data, employ an unsupervised machine learning pipeline to deconvolute overlapping bands and identify chemically significant features.
- Algorithm Workflow:
- Pre-process spectra (e.g., baseline correction, normalization).
- Apply Principal Component Analysis (PCA) for dimensionality reduction.
- Use Density-Based Spatial Clustering (DBSCAN) to group spectra based on intrinsic spectral features, effectively separating signals from spectral noise and artifacts [77].
Protocol 2: Identifying and Reporting Impurities in ¹H NMR for Small Molecule Analysis
This protocol provides a systematic approach to distinguishing impurities from the target compound, which is critical for pharmaceutical R&D [76] [71] [79].
1. Sample Preparation:
- Dissolve approximately 5-10 mg of the sample in 0.6 mL of an appropriate deuterated solvent (e.g., CDCl₃, DMSO-d₆).
- Transfer the solution to a clean, dry NMR tube.
2. Data Acquisition (300 MHz Spectrometer Example):
- Experiment: Standard ¹H NMR
- Pulse Program: zg30
- Number of Scans: 16-32
- Relaxation Delay (D1): 1 second
- Spectral Width: 20 ppm
3. Data Interpretation and Impurity Identification:
- Analyze the spectrum for signals that do not integrate to whole-number ratios of protons relative to the main compound.
- Compare all singlets, particularly those integrating for less than one proton, against known NMR chemical shift tables for common impurities and residual solvents [76].
- Confirmatory 2D NMR: If an impurity signal is suspected but cannot be identified from 1D data alone, acquire 2D NMR data (e.g., COSY, HSQC, HMBC). A signal that shows no correlations in a 2D experiment likely belongs to an impurity and not the target compound [71] [79].
- Reporting: For regulatory compliance, document the identity and level of any impurity observed. NMR is orthogonal to LC-MS and excels at detecting isomeric impurities and non-ionizable compounds [71].
Protocol 3: Managing Concentration Effects in FT-IR Analysis of Protein Formulations
This protocol leverages recent advancements in ATR-FT-IR imaging for in-line monitoring of biopharmaceuticals, which often require analysis at high concentrations [75].
1. Experimental Setup:
- Utilize an ATR-FT-IR spectrometer equipped with a microfluidic channel flow cell.
- The system allows for control of protein concentration, pH, and temperature, mimicking process conditions.
2. In-Line Data Acquisition:
- Flow the protein formulation (e.g., a monoclonal antibody at ~200 mg/mL) through the microfluidic channel.
- Instrumental Settings:
- Collect spectra continuously or at set intervals (e.g., every 30 seconds).
- Use a resolution of 4-8 cm⁻¹.
- Co-add a sufficient number of scans to maintain a good signal-to-noise ratio despite the flow condition.
- The ATR technique is not limited by protein concentration, making it ideal for analyzing highly concentrated mAb formulations used in subcutaneous injections [75].
3. Data Analysis for Stability Assessment:
- Monitor the Amide I (~1650 cm⁻¹) and Amide II (~1550 cm⁻¹) bands for changes in shape and intensity.
- Shifts to higher wavenumbers in the Amide I band can indicate protein unfolding or changes in secondary structure.
- The multi-channel design of advanced setups allows for direct comparison of formulations under different conditions, reducing experimental variability and providing high-throughput data [75].
Visualized Workflows and Signaling Pathways
The following diagrams summarize the logical workflows for addressing the key sample-related issues discussed in this document.
Fourier-Transform Infrared (FTIR) and Nuclear Magnetic Resonance (NMR) spectroscopy are cornerstone techniques for functional group identification and structural elucidation in pharmaceutical research and development. While traditional analysis focuses on interpreting key peaks or shifts, advanced data processing techniques unlock significantly more information from complex spectral data. Chemometrics—the application of mathematical and statistical methods to chemical data—and spectral deconvolution are powerful approaches that transform spectra into quantitative, actionable information about complex mixtures and molecular structures.
These advanced techniques address critical challenges in modern drug development, including the analysis of multi-component formulations, detection of trace impurities, and monitoring of reaction pathways. By moving beyond qualitative assessment, researchers can achieve greater accuracy in identifying functional groups, quantifying active pharmaceutical ingredients (APIs), and validating product composition.
Core Chemometric Methods for Spectral Analysis
Multivariate Calibration and Pattern Recognition
Chemometric methods enable researchers to extract meaningful chemical information from complex, overlapping spectral signals. Two fundamental approaches are widely used for quantitative analysis and pattern recognition:
Partial Least Squares (PLS) Regression: PLS is a multivariate calibration technique that establishes a relationship between spectral variables (e.g., absorbance at different wavenumbers) and chemical properties (e.g., concentration). It is particularly effective when predictor variables are numerous and highly correlated, as is common in FTIR and NMR spectra. PLS models can deconvolute spectra even when differences between components are minute, making them invaluable for analyzing co-eluting compounds in chromatographic separations monitored by spectroscopy [80] [81]. The method works by projecting the predictive and observed variables onto a new space defined by latent variables (factors) that maximize covariance.
Principal Component Analysis (PCA): PCA is an unsupervised pattern recognition method used for exploratory data analysis. It reduces the dimensionality of spectral data by transforming original variables into a smaller set of principal components (PCs) that capture the greatest variance in the dataset. In practice, PCA can reveal natural clustering of samples, identify outliers, and highlight key spectral features responsible for variance between samples. For example, PCA has been applied to mid-IR spectra of ibuprofen in supercritical CO₂ to determine changes in spectral contributions from different molecular conformers under varying thermodynamic conditions [82].
Multi-Way Analysis and Machine Learning Approaches
Beyond traditional two-dimensional data analysis, advanced techniques have emerged for handling more complex data structures:
PARAFAC (Parallel Factor Analysis): PARAFAC is a multi-way decomposition method particularly suited for analyzing three-dimensional data arrays (e.g., excitation-emission matrices, spectral-pH time series). Unlike PCA, PARAFAC provides a unique solution without rotational ambiguity, making it ideal for spectral deconvolution. It has been successfully applied to UV spectrophotometric data of acetylsalicylic acid and ascorbic acid mixtures, enabling spectral deconvolution, estimation of pKa values, and detection of potential decomposition products like salicylic acid [80].
Artificial Neural Networks (ANNs): Machine learning approaches, particularly ANNs, have shown remarkable success in spectral interpretation. By training on large spectral databases, ANNs can learn to identify functional groups with high accuracy. Research demonstrates that models trained simultaneously on multiple spectroscopic data types (FT-IR, ¹H NMR, and ¹³C NMR) significantly outperform single-technique models, achieving a macro-average F1 score of 0.93 for identifying 17 different functional groups [5]. Recent transformer models can even predict complete molecular structures directly from IR spectra when combined with chemical formula information, achieving top-1 accuracy of 44.4% for compounds containing 6-13 heavy atoms [4].
Table 1: Key Chemometric Methods and Their Applications in FTIR and NMR Analysis
| Method | Type | Primary Application | Advantages | Limitations |
|---|---|---|---|---|
| PLS Regression | Multivariate calibration | Quantitative analysis of multi-component mixtures | Handles correlated variables; works with noisy data | Requires calibration set; model transfer challenging |
| PCA | Unsupervised pattern recognition | Exploratory data analysis; outlier detection | No prior knowledge required; reduces data dimensionality | Results sometimes difficult to interpret physically |
| PARAFAC | Multi-way decomposition | Spectral deconvolution; analysis of complex mixtures | Unique solution; physically interpretable components | Requires multi-way data; sensitive to outliers |
| ANN | Machine learning | Functional group identification; structure prediction | High accuracy; handles complex patterns | Large training data required; "black box" nature |
Spectral Deconvolution Techniques
Fundamentals of Spectral Deconvolution
Spectral deconvolution is the process of separating overlapping bands in spectra to identify individual components, quantify their contributions, and determine their parameters (position, intensity, width). This is particularly crucial in the fingerprint region of FTIR spectra (400-1500 cm⁻¹) where complex, molecule-specific patterns appear, and in NMR spectra where signal overlap occurs in crowded regions.
The foundation of many quantitative deconvolution approaches is the Lambert-Beer law, which states that absorbance is proportional to concentration, pathlength, and a compound-specific molar absorptivity. Custom algorithms leveraging this principle can deconvolute absorbance data autonomously into component concentrations and their respective spectra without requiring offline analytical calibration [81].
Advanced Deconvolution Approaches
Two-Dimensional Correlation Spectroscopy (2D-COS): 2D-COS analyzes spectral intensity changes under external perturbation (e.g., temperature, concentration, pH) to reveal correlations between different spectral regions. This technique enhances spectral resolution by spreading peaks along a second dimension and establishes sequential order of spectral changes. Applied to mid-IR spectra of ibuprofen in supercritical CO₂, 2D-COS revealed correlations between spectral contributions from different conformers and showed how their relative intensities change with temperature [82].
Curve Fitting and Band Analysis: This approach involves mathematically fitting experimental spectra with multiple component peaks (typically Gaussian, Lorentzian, or mixed functions). The process includes identifying the number of underlying components, estimating their initial parameters, and iteratively optimizing the fit. This technique is particularly valuable for quantifying the relative abundance of different molecular conformations or analyzing complex band systems in both FTIR and NMR spectra.
Table 2: Spectral Deconvolution Techniques for FTIR and NMR Analysis
| Technique | Principle | Data Requirements | Typical Applications |
|---|---|---|---|
| Curve Fitting | Mathematical fitting of spectra with multiple component peaks | High-quality spectra with partially resolved bands | Conformational analysis; crystallinity studies |
| 2D-COS | Analysis of spectral changes under external perturbation | Time-series or variable-condition spectra | Determining transition sequences; identifying coupled bands |
| Derivative Spectroscopy | Computation of 1st or 2nd derivatives of spectra | High signal-to-noise ratio spectra | Enhancing resolution of overlapping bands; background elimination |
| Self-Modeling Curve Resolution | Extraction of pure components without prior information | Spectral series of mixtures with varying composition | Tracking reaction pathways; impurity profiling |
Experimental Protocols
Protocol 1: PLS Regression for Quantitative Analysis of API in Formulations
This protocol describes the development of a PLS model for quantifying active pharmaceutical ingredients using FTIR spectroscopy, adapted from studies on azodicarboxamide analysis [55].
Materials and Reagents:
- FTIR spectrometer with ATR accessory
- Pure API reference standard
- Excipients (representative of formulation)
- Appropriate solvent (e.g., DMSO for azodicarboxamide [55])
- Analytical balance (±0.1 mg accuracy)
Procedure:
- Standard Preparation: Prepare a stock solution of API reference standard at known concentration. Serially dilute to create calibration standards covering the expected concentration range (e.g., 10-40 mg/mL for azodicarboxamide [55]).
- Sample Preparation: For solid dosage forms, homogenize tablets/capsules and prepare solutions in appropriate solvent. For direct analysis, ensure consistent particle size and packing for ATR measurements.
- Spectral Acquisition: Collect FTIR spectra of all standards and samples using consistent parameters (resolution: 4-8 cm⁻¹, scans: 16-64, range: 4000-400 cm⁻¹). Include solvent/background spectra for subtraction.
- Data Preprocessing: Apply vector normalization, baseline correction, and spectral derivatives if needed to enhance spectral features.
- Model Development:
- Divide data into calibration (≥70%) and validation sets
- Select optimal spectral regions for analysis (e.g., 2000-1500 cm⁻¹ for azodicarboxamide [55])
- Use cross-validation to determine optimal number of latent variables
- Build PLS model relating spectral data to reference concentrations
- Model Validation: Assess model performance using root mean square error of calibration (RMSEC), root mean square error of prediction (RMSEP), and correlation coefficients (R²). For azodicarboxamide, linear ranges with correlation coefficients of 0.9998 have been achieved [55].
Protocol 2: PARAFAC for Spectral Deconvolution of Multi-Component Mixtures
This protocol applies PARAFAC to deconvolute overlapping spectral signals from mixtures, based on methods used for acetylsalicylic acid and ascorbic acid analysis [80].
Materials and Reagents:
- UV-Vis or FTIR spectrometer
- Pure components (reference standards)
- Buffer solutions covering pH range (e.g., pH 1.0-5.5)
- Temperature-controlled sample compartment
Procedure:
- Experimental Design: Prepare mixtures with systematically varied concentrations of all components. For pH-dependent studies, prepare solutions across relevant pH range (e.g., 1.0-5.5 [80]).
- Spectral Acquisition: Collect full spectra for all mixture combinations. Maintain consistent measurement conditions (slit widths, scanning speed, temperature).
- Data Array Construction: Arrange spectra into three-dimensional array (samples × wavelengths × pH, or samples × wavelengths × concentration).
- PARAFAC Decomposition:
- Preprocess data (normalization, baseline correction)
- Determine appropriate number of components via core consistency diagnostic
- Apply PARAFAC algorithm to decompose array into three matrices: concentrations, spectra, and additional parameter (e.g., pH profile)
- Validate model using split-half analysis or residual analysis
- Interpretation: Examine extracted spectral profiles and compare to reference standards. Use concentration profiles for quantitative analysis. For pharmaceutical mixtures, this approach has achieved recovery rates between 97.6-103.6% [80].
Protocol 3: Machine Learning for Functional Group Identification
This protocol outlines the development of an artificial neural network for functional group identification from combined FTIR and NMR spectra, based on recent advances in spectroscopic machine learning [5] [4].
Materials and Software:
- FTIR and NMR spectrometers
- Spectral database (e.g., NIST for FTIR, SDBS for NMR)
- Programming environment (Python, R)
- Machine learning libraries (TensorFlow, PyTorch, scikit-learn)
Procedure:
- Data Collection: Compile FTIR, ¹H NMR, and ¹³C NMR spectra for diverse compounds with known structures. Sources include public databases (NIST Chemistry WebBook, SDBS) [5].
- Data Preprocessing:
- Model Architecture: Implement artificial neural network with:
- Input layers for each spectral type
- Hidden layers with activation functions (ReLU, sigmoid)
- Output layer with sigmoid activation for multi-label classification
- Model Training:
- Apply stratified k-fold cross-validation (e.g., 5-fold)
- Use binary cross-entropy loss function
- Optimize with adaptive moment estimation (Adam)
- Validation: Evaluate performance using F1 scores, precision, recall. Integrated FTIR-NMR models have achieved macro-average F1 scores of 0.93 for functional group identification [5].
Data Presentation and Analysis
Quantitative Performance of Chemometric Methods
Table 3: Performance Metrics of Advanced Data Processing Techniques in Pharmaceutical Analysis
| Application | Technique | Performance Metrics | Reference Compound |
|---|---|---|---|
| API Quantification | PLS (FTIR) | Linear range: 10-40 mg/mL; R²: 0.9998; n=15 | Azodicarboxamide [55] |
| Impurity Analysis | Transmission FTIR | LOD: 5.0 μg/g; LOQ: 15 μg/g; R² >0.9994; n=18 | Azodicarboxamide impurities [55] |
| Multi-Component Analysis | PARAFAC (UV) | Recovery: 97.6-103.6%; pKa estimation | Acetylsalicylic acid, Ascorbic acid [80] |
| Functional Group ID | ANN (FTIR+NMR) | Macro F1-score: 0.93; 17 functional groups | Diverse organic compounds [5] |
| Structure Elucidation | Transformer (FTIR) | Top-1 accuracy: 44.4%; Top-10: 69.8% | Compounds with 6-13 heavy atoms [4] |
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 4: Key Research Reagent Solutions for Advanced Spectroscopic Analysis
| Reagent/Material | Function/Application | Notes/Specifications |
|---|---|---|
| DMSO-d₆ | NMR solvent for polar compounds | Minimizes exchangeable protons; suitable for a wide range of pharmaceuticals |
| CDCl₃ | NMR solvent for non-polar compounds | Maintains consistency for database matching; tetramethylsilane (TMS) as internal standard [5] |
| KBr Pellet | FTIR sample preparation for solids | Provides transparent matrix for transmission measurements [83] |
| ATR Crystals | FTIR accessory for minimal sample prep | Diamond, ZnSe, or germanium elements with high refractive index [84] |
| Reference Standards | Method calibration and validation | High-purity compounds for building calibration models [55] |
| Buffer Solutions | pH-controlled studies | Phosphate, citrate, or acetate buffers for pH-dependent spectral analysis [80] |
Workflow Visualization
Advanced Spectral Data Processing Workflow This diagram illustrates the comprehensive workflow for applying chemometrics and spectral deconvolution to FTIR and NMR data, from sample preparation through method selection to final validation.
Advanced data processing techniques have transformed FTIR and NMR spectroscopy from qualitative identification tools to powerful quantitative and diagnostic platforms in pharmaceutical research. Chemometric methods like PLS regression and PARAFAC enable precise quantification of active ingredients and impurities while effectively deconvoluting complex spectral mixtures. Machine learning approaches further enhance these capabilities by enabling automated functional group identification and even complete structure elucidation directly from spectral data.
The integration of these computational methods with traditional spectroscopic analysis creates a robust framework for addressing challenging problems in drug development, including formulation analysis, impurity profiling, and reaction monitoring. As these techniques continue to evolve, particularly with advances in artificial intelligence and multi-way analysis, they will play an increasingly vital role in accelerating pharmaceutical development while ensuring product quality and consistency.
Validating Spectral Interpretations and Avoiding Common Misinterpretations
Fourier Transform Infrared (FT-IR) and Nuclear Magnetic Resonance (NMR) spectroscopy are cornerstone techniques in modern molecular analysis, providing critical insights into functional group identity, molecular structure, and dynamics. FT-IR spectroscopy measures molecular vibrations through the absorption of infrared light, generating a spectrum that serves as a molecular "fingerprint" [85]. NMR spectroscopy exploits the magnetic properties of certain nuclei, such as ¹H and ¹³C, to provide detailed information on the chemical environment, connectivity, and conformation of molecules [86]. The power of these techniques is magnified when they are used in concert, as they offer complementary data for structural elucidation. However, the accuracy of any conclusion drawn hinges on the rigorous validation of spectral interpretations and a clear understanding of common pitfalls that can lead to misinterpretation. This application note provides a structured framework, within the context of functional group research, to help researchers and drug development professionals navigate these challenges effectively.
Core Principles and Instrumentation
FT-IR Spectroscopy Fundamentals
In FT-IR spectroscopy, when IR radiation interacts with a sample, specific frequencies are absorbed that correspond to the vibrational energies of molecular bonds, such as stretching, bending, or twisting [85]. The fundamental output is an interferogram, which is transformed via a Fast Fourier Transform (FFT) algorithm into a familiar intensity-versus-wavenumber (cm⁻¹) spectrum [85]. The absorption of IR radiation requires a change in dipole moment, making polar bonds (e.g., C=O, O–H, N–H) particularly strong IR absorbers [85].
Modern FT-IR instruments support several sampling geometries, each suited to different sample types:
- Transmission: IR light passes through a thin film or pellet; requires careful thickness control [85].
- Attenuated Total Reflectance (ATR): The most popular modern technique, it uses an internal reflection element (e.g., diamond) to analyze surfaces of solids, liquids, and gels with minimal preparation [85].
- Diffuse Reflectance (DRIFTS): Ideal for collecting scattered radiation from powders or rough surfaces [85].
NMR Spectroscopy Fundamentals
NMR spectroscopy provides information based on the interaction of atomic nuclei with an external magnetic field. The precise resonance frequency of a nucleus—its chemical shift (δ)—is exquisitely sensitive to its local electronic environment, making it a powerful probe for functional group identification and molecular structure [86]. The dimensionless chemical shift parameter allows for comparisons independent of the spectrometer's magnetic field strength [86]. Furthermore, NMR can probe scalar J-couplings between nuclei, revealing connectivity and stereochemistry.
Computational methods, particularly Density Functional Theory (DFT), have revolutionized NMR by enabling accurate prediction of NMR parameters like chemical shifts and coupling constants. This allows for direct comparison between experimental data and quantum-mechanically simulated spectra, providing a powerful method for structural verification [86].
Experimental Protocols
Protocol for FT-IR Analysis Using ATR
This protocol is adapted for the ubiquitous ATR sampling technique, which requires minimal sample preparation [85] [8].
- Instrument Preparation: Power on the FT-IR spectrometer and allow it to warm up and stabilize. Ensure the ATR crystal (e.g., diamond, ZnSe) is meticulously clean. Purge the optical compartment with dry nitrogen to minimize spectral contributions from atmospheric water vapor and CO₂ [85].
- Background Acquisition: Collect a background spectrum (also called a reference spectrum) with a clean ATR crystal, under the same instrumental conditions (resolution, number of scans) that will be used for the sample. This records the instrument and environmental signature, which will be automatically subtracted from the sample spectrum [85].
- Sample Loading: For solids, place a finely ground portion of the sample directly onto the ATR crystal. For liquids, deposit a small droplet. Use the instrument's pressure clamp to ensure firm, even contact between the sample and the crystal. Inadequate contact is a common source of weak, distorted spectra [85].
- Spectral Acquisition: Collect the sample spectrum. A resolution of 4 cm⁻¹ is typically sufficient for most chemical analyses [85]. The number of scans is often set to 32 or 64 as a balance between signal-to-noise ratio and acquisition time [87].
- Data Processing: Apply necessary post-processing steps, which may include:
- Baseline Correction: To correct for any sloping or curved baseline.
- Atmospheric Subtraction: To remove any residual water vapor or CO₂ bands.
- Normalization: To enable comparison between different samples.
Protocol for ¹H-NMR Analysis of Small Molecules
This protocol outlines the steps for a standard ¹H-NMR experiment in solution [87] [88].
- Sample Preparation: Dissolve approximately 1-10 mg of the purified compound in 0.5-0.7 mL of a deuterated solvent (e.g., CDCl₃, D₂O, DMSO-d₆). The deuterated solvent provides a signal for the instrument lock system. Transfer the solution to a clean, dry NMR tube.
- Instrument Setup: Insert the NMR tube into the spectrometer. Tune and match the probe to the sample. Lock the magnetic field to the deuterium signal of the solvent. Shim the magnet to optimize field homogeneity and achieve good spectral resolution.
- Acquisition Parameters: Set the acquisition parameters. A standard ¹H-NMR experiment typically uses:
- Data Acquisition and Processing: Run the experiment. After acquisition, process the Free Induction Decay (FID) by applying:
- Fourier Transformation: Converts the time-domain FID to a frequency-domain spectrum.
- Phase Correction: To obtain pure absorption-mode peaks.
- Baseline Correction.
- Referencing: Calibrate the spectrum by setting the known solvent residual peak to its standard chemical shift value (e.g., CHCl₃ in CDCl₃ to 7.26 ppm).
Validation of Spectral Interpretations
Cross-Technique Corroboration
The most robust validation strategy involves cross-referencing data from multiple analytical techniques. A functional group identified by a characteristic C=O stretch in FT-IR around 1700-1750 cm⁻¹ should be corroborated by the observation of the corresponding carbon signal in a ¹³C-NMR spectrum in the δ 160-220 ppm region [88]. Similarly, an O-H stretch in FT-IR (3200-3600 cm⁻¹) should align with a broad, exchangeable proton signal in the ¹H-NMR spectrum.
Computational Validation
Table 1: Computational Methods for Spectral Validation
| Method | Description | Application in Validation |
|---|---|---|
| Quantum Chemical (DFT) | Predicts NMR parameters (chemical shifts, J-couplings) and IR vibrational frequencies from first principles [86]. | Simulated spectra are directly compared to experimental data to verify proposed structures. Discrepancies can indicate an incorrect assignment. |
| Machine Learning (ML) | Uses algorithms trained on large spectral databases to predict spectra or automate spectral assignment [86]. | Accelerates the assignment process for complex molecules and helps identify patterns that may be missed manually. |
| Spectral Databases | Commercial and public libraries of reference spectra for known compounds. | Provides a direct empirical comparison. The unknown spectrum should closely match the reference spectrum of the proposed compound. |
Quantitative and Statistical Validation
For quantitative analysis, ensure the system obeys the Beer-Lambert law (for FT-IR) by demonstrating a linear relationship between absorbance and concentration [85]. Employ chemometric techniques like Principal Component Analysis (PCA) or Partial Least Squares (PLS) modeling to extract meaningful information from complex spectral data and build robust, multivariate calibration models [8]. Method validation for quantitative assays should include assessments of reproducibility, precision, and sensitivity [85].
Common Pitfalls and How to Avoid Them
Table 2: Common Spectral Misinterpretations and Preventive Strategies
| Pitfall | Consequence | Preventive Strategy |
|---|---|---|
| Inadequate Background/Reference | Strong atmospheric water vapor (~3500 cm⁻¹, ~1650 cm⁻¹) and CO₂ (~2350 cm⁻¹) bands obscure sample peaks [85]. | Always collect a fresh background under identical conditions. Consistently purge the instrument with dry nitrogen [85]. |
| Poor Sample Preparation | In ATR, uneven contact causes distorted bands. In NMR, poor shimming or paramagnetic impurities lead to broad peaks. | Ensure homogeneous sample and firm ATR contact. For NMR, use filtered, high-purity samples and proper shimming. |
| Overlooking Solvent/Sample Interactions | Solvent can affect chemical shifts and hydrogen bonding, altering spectral appearance. | Note the solvent used in both FT-IR and NMR. Be aware of solvent peaks in NMR and water absorption in FT-IR. |
| Over-Interpretation of Small Peaks | Assigning minor impurities or artifacts as key functional groups. | Correlate peak intensities with expected stoichiometry. Use 2D-NMR experiments (e.g., COSY, HSQC) to confirm connectivity. |
| Ignoring Concentration Effects | High concentration can cause hydrogen bonding and peak broadening/shifting, especially for O-H and N-H stretches. | Analyze samples at different concentrations to identify concentration-dependent effects. |
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Reagents and Materials for FT-IR and NMR Analysis
| Item | Function/Brief Explanation |
|---|---|
| Deuterated Solvents (e.g., CDCl₃, DMSO-d₆) | Provides a non-protonated medium for NMR analysis and a deuterium signal for the instrument lock system [87]. |
| ATR Crystals (Diamond, ZnSe, Ge) | The Internal Reflection Element (IRE) in ATR-FT-IR. Material choice balances durability, refractive index, and spectral range [85]. |
| Potassium Bromide (KBr) | Used for preparing pellets for transmission FT-IR analysis of solid samples. |
| NMR Tube | A high-precision glass tube designed to hold the sample and withstand spinning within the NMR magnet. |
| Internal Standard (e.g., TMS) | Provides a reference peak (δ 0.00 ppm) for chemical shift calibration in NMR spectroscopy. |
| Calibration Standards | Known compounds for verifying wavelength/wavenumber (FT-IR) and chemical shift (NMR) accuracy of the instrument. |
Workflow and Pathway Visualizations
Spectral Validation Workflow
The following diagram illustrates a systematic workflow for validating spectral interpretations, integrating both experimental and computational approaches.
FT-IR Sampling Techniques
This diagram summarizes the key sampling techniques available in modern FT-IR spectroscopy and their typical applications.
Application in Drug Development: A Case Study
The integrated use of FT-IR and NMR is powerfully illustrated in the development and characterization of novel Methyl α-D-glucopyranoside (MGP) derivatives with antimicrobial and anticancer potential [88]. In this study:
- FT-IR spectroscopy was used to confirm the successful introduction of new functional groups, such as acyl chains, by identifying their characteristic vibrational bands (e.g., C=O stretch) [88].
- ¹H- and ¹³C-NMR spectroscopy provided definitive evidence of the chemical structure and the specific sites of substitution on the sugar ring, allowing researchers to distinguish between different regioisomers [88].
The correlation of data from both techniques was essential for verifying the synthetic products before biological testing. Furthermore, the study extended to in silico methods, using molecular docking and predictions of activity spectra for substances (PASS) to validate the biological relevance of the interpreted structures, showcasing a modern, multi-technique approach to drug development [88].
The reliable interpretation of FT-IR and NMR spectra is a critical skill in functional group research and drug development. This reliability is achieved not by a single test, but through a systematic validation strategy. This strategy encompasses meticulous sample preparation, diligent control of experimental conditions, and, most importantly, the synergistic use of multiple analytical and computational techniques. By adhering to the protocols, being aware of common pitfalls, and leveraging the workflows outlined in this document, researchers can significantly enhance the confidence in their spectral assignments, thereby accelerating the path from spectral data to meaningful scientific conclusions.
Synergistic Power: Choosing and Combining FTIR and NMR for Definitive Analysis
Within the framework of qualitative analysis for functional group research, Fourier-Transform Infrared (FTIR) spectroscopy and Nuclear Magnetic Resonance (NMR) spectroscopy serve as cornerstone analytical techniques. The selection between them hinges on a clear understanding of their respective information output, sensitivity, and operational requirements. FTIR spectroscopy probes molecular vibrational energy levels, providing characteristic fingerprints for specific chemical bonds and functional groups [89]. In contrast, NMR spectroscopy exploits the magnetic properties of certain nuclei, such as ^1^H, placed in a static magnetic field, yielding detailed information about the molecular structure, including the specific chemical environment of atoms and their connectivity [90] [40]. This application note provides a direct comparison of these two powerful techniques, supplemented with detailed experimental protocols from relevant food science applications, to guide researchers and drug development professionals in their analytical strategy.
Technical Comparison: FTIR vs. NMR
The following table summarizes the core technical characteristics of FTIR and NMR spectroscopy, highlighting their complementary strengths and weaknesses.
Table 1: Direct comparison of FTIR and NMR spectroscopy for functional group analysis.
| Parameter | FTIR Spectroscopy | NMR Spectroscopy |
|---|---|---|
| Fundamental Principle | Measures absorption of IR radiation, causing changes in molecular dipole moment and vibrational energy levels [89] | Measures absorption of radiofrequency radiation by atomic nuclei (e.g., ^1^H, ^13^C) in a magnetic field, causing nuclear spin transitions [90] |
| Primary Information Output | Identification of functional groups and chemical bonds; molecular "fingerprint" [89] | Quantitative molecular structure elucidation; chemical environment, connectivity, and dynamics [40] |
| Sensitivity | High for polar bonds; suitable for bulk sample analysis | Inherently lower than FTIR; requires more sample or longer acquisition times, though field strength is a major factor [90] |
| Sample Throughput | Very High (acquisition in minutes) [89] | Moderate to Low (acquisition can take 10+ minutes to hours) [90] |
| Sample Form | Gas, liquid, solid (minimal preparation) [89] | Primarily liquid (requires dissolution in deuterated solvent) [90] |
| Key Quantitative Strengths | Good for functional group quantification using Beer-Lambert law [91] | Excellent for quantification (qNMR); signal intensity is directly proportional to nucleus count [40] |
| Key Limitations | Peak overlap in complex mixtures; less specific for detailed structure | Low sensitivity; requires deuterated solvents; instrument cost and complexity [90] |
A notable comparative study on detecting hazelnut oil adulteration in olive oil demonstrated that bench-top 60 MHz ^1^H NMR performed at least as well as FTIR in terms of sensitivity, with a detection limit of 11.2% w/w, and offered improved specificity, making it a superior screening tool for this particular application [90].
Experimental Protocols for Direct Comparison
To illustrate the practical application of both techniques in a single research context, the following protocols are adapted from studies on edible oil authentication and starch esterification.
Protocol 1: Detection of Oil Adulteration using FTIR and NMR
This protocol simulates the adulteration of olive oil with hazelnut oil and outlines parallel analysis using both techniques [90].
Research Reagent Solutions & Materials
Table 2: Essential reagents and materials for the oil adulteration study.
| Item | Function / Specification |
|---|---|
| Extra Virgin Olive Oil | High-value matrix to be tested for adulteration. |
| Hazelnut Oil | Model adulterant due to its similar fatty acid profile. |
| Chloroform (non-deuterated) | NMR sample preparation: reduces oil viscosity and provides a chemical shift reference [90]. |
| FTIR Spectrometer | Equipped with a liquid cell accessory. |
| Benchtop NMR Spectrometer | 60 MHz ^1^H NMR (e.g., Pulsar). |
Sample Preparation
- Mixture Preparation: Prepare independent calibration (Batch 1) and test (Batch 2) samples by gravimetrically mixing hazelnut oil into olive oil in the range of 4–26% w/w to simulate adulteration [90].
- NMR Sample: For each sample, mix approximately 2 mL of the oil mixture with 2 mL of non-deuterated chloroform in a standard 5 mm NMR tube [90].
- FTIR Sample: Apply a small volume of the neat oil mixture directly onto the crystal of an Attenuated Total Reflectance (ATR) accessory or load into a suitable liquid cell [90].
Data Acquisition
- NMR Acquisition:
- Instrument: 60 MHz benchtop ^1^H NMR spectrometer.
- Acquisition Time: ~10 minutes per sample.
- The chloroform peak is used to reference the chemical shift scale [90].
- FTIR Acquisition:
- Instrument: FTIR spectrometer with MIR source.
- Spectral Range: 4000–400 cm^-1.
- Acquisition Time: ~10 minutes per sample (including background scan) [90].
Data Analysis
- NMR Analysis: Employ a whole-spectrum chemometric approach (e.g., Principal Component Analysis or Partial Least Squares regression) on the preprocessed ^1^H NMR spectra to build a model for detecting and quantifying the adulteration level [90].
- FTIR Analysis: Similarly, apply chemometric models to the FTIR spectral data, particularly focusing on the fingerprint region, to predict the concentration of the adulterant [90].
The workflow for this comparative analysis is outlined below.
Protocol 2: Determining the Degree of Substitution in Starch Esters
This protocol describes the use of FTIR for validation and ^1^H NMR as a reference method for determining the Degree of Substitution (DS) in acylated starch [91].
Research Reagent Solutions & Materials
- Starch: Rice or quinoa starch.
- Acylating Reagents: Acetic, propionic, or butyric anhydride.
- Sodium Hydroxide (NaOH) Solution: 0.7 M, for pH control during reaction.
- Deuterated Dimethyl Sulfoxide (DMSO-d₆): Solvent for ^1^H NMR analysis.
- Potassium Bromide (KBr): For preparing solid pellets for FTIR (if not using ATR).
Starch Ester Synthesis
- Disperse 70.0 g of starch in 280 g of distilled water with stirring.
- Adjust the pH to 8.5 ± 0.2 with 0.7 M NaOH.
- Gradually add the desired fatty acid anhydride (e.g., acetic anhydride) while maintaining the pH at 8.5 with NaOH.
- Once the reaction is complete (pH stabilizes), adjust the pH to 6.5.
- Isolate the product by centrifugation, wash with water and acetone, and dry at room temperature [91].
DS Determination by Stoichiometry
The DS can be calculated directly from the molar consumption of NaOH during the reaction, which is monitored by the mass of NaOH solution consumed [91].
DS Determination by FTIR
- Sample Preparation: Prepare a solid sample pellet using KBr or use the ATR method on the dry, powdered starch ester.
- Data Acquisition: Acquire the FTIR spectrum in the range of 4000–400 cm^-1.
- Data Analysis: Confirm the success of esterification by the appearance of the characteristic carbonyl (C=O) stretch band at ~1700 cm⁻¹ and the C–O stretch at ~1200 cm⁻¹. The intensity of these bands can be used for quantitative DS determination based on the Beer-Lambert law [91].
DS Determination by ^1^H NMR
- Sample Preparation: Dissolve the starch ester in DMSO-d₆.
- Data Acquisition: Acquire a quantitative ^1^H NMR spectrum.
- Data Analysis: Identify the protons from the acyl group (e.g., the methyl protons of an acetate group at ~1.8–2.1 ppm) and the anhydroglucose unit protons. The DS is calculated from the integral ratio between the acyl group protons and the sugar ring protons [91].
The logical relationship between these analytical techniques in the starch study is shown in the following diagram.
FTIR and NMR spectroscopy offer complementary capabilities for functional group research and qualitative analysis. FTIR excels as a rapid, high-throughput fingerprinting technique for identifying functional groups with minimal sample preparation. In contrast, NMR provides unparalleled detail on molecular structure and quantification, albeit with greater sample and solvent requirements. The choice between them is not a matter of superiority but of strategic application. For rapid screening and functional group confirmation, FTIR is often the optimal first step. For definitive structural elucidation and precise quantification in complex mixtures, NMR is the definitive tool. As demonstrated in the cited protocols, their combined use provides a powerful, orthogonal analytical approach that delivers robust and comprehensive results for researchers in drug development and beyond.
In the field of molecular analysis, particularly for functional group research and drug development, Fourier Transform Infrared (FTIR) and Nuclear Magnetic Resonance (NMR) spectroscopy stand as two foundational techniques. While both provide critical insights into molecular structure, they operate on fundamentally different principles and are optimized for distinct analytical scenarios. FTIR spectroscopy measures the absorption of infrared light by molecular bonds, resulting in a spectrum that serves as a unique molecular fingerprint based on vibrational energies [92] [14]. In contrast, NMR spectroscopy exploits the magnetic properties of specific atomic nuclei (such as ^1H or ^13C) when placed in a strong magnetic field, providing detailed information about the carbon-hydrogen framework of organic compounds [5] [93]. The selection between these techniques is not merely a matter of preference but a strategic decision that significantly impacts the quality, depth, and applicability of analytical results in research and development settings.
For researchers and drug development professionals, understanding the complementary strengths and limitations of FTIR and NMR is crucial for designing efficient analytical workflows. This application note provides a structured decision matrix and detailed experimental protocols to guide this selection process, framed within the context of functional group identification and structural elucidation. By synthesizing current research and applications, we establish a framework for leveraging these techniques individually and in combination to address complex analytical challenges across pharmaceutical, environmental, and materials science domains.
Technical Comparison: Operating Principles and Analytical Capabilities
Fundamental Principles and Measurable Parameters
FTIR spectroscopy functions on the principle that chemical bonds vibrate at specific frequencies when exposed to infrared radiation, with these vibrations being highly sensitive to molecular structure and environment [92] [14]. The resulting spectrum plots absorbance or transmittance against wavenumber (typically covering 4000-400 cm⁻¹), providing information about the functional groups present in a sample. Different regions of the FTIR spectrum correspond to specific types of molecular vibrations: the single-bond region (4000-2500 cm⁻¹) captures O-H, N-H, and C-H stretching; the triple-bond region (2500-2000 cm⁻¹) identifies C≡C and C≡N groups; the double-bond region (2000-1500 cm⁻¹) is dominated by C=O and C=C stretching; while the fingerprint region (1500-500 cm⁻¹) contains complex patterns unique to individual compounds [14].
NMR spectroscopy, conversely, relies on the absorption of radiofrequency radiation by atomic nuclei with non-zero spin (most commonly ^1H and ^13C) when placed in a strong external magnetic field [5] [93]. The precise resonance frequency of these nuclei depends on their local electronic environment, providing detailed information about molecular structure, including the connectivity of atoms, stereochemistry, and dynamics. Proton NMR (^1H NMR) reveals information about hydrogen atoms in different chemical environments, while carbon-13 NMR (^13C NMR) provides complementary data about the carbon skeleton [5]. Two-dimensional NMR techniques can further elucidate through-bond and through-space connections between atoms, offering unparalleled insights into molecular architecture.
Comparative Performance Metrics
Table 1: Analytical Capabilities of FTIR and NMR Spectroscopy
| Parameter | FTIR Spectroscopy | NMR Spectroscopy |
|---|---|---|
| Primary Information | Functional group identification, molecular fingerprints | Molecular structure, atomic connectivity, quantitative analysis |
| Sample Throughput | High (typically minutes per sample) | Moderate to low (typically minutes to hours per sample) |
| Quantitative Capability | Semi-quantitative with calibration | Excellent quantitative accuracy (qNMR) [94] [95] |
| Sensitivity | Moderate | Moderate to high (dependent on magnetic field strength) |
| Sample Requirements | Minimal preparation; solids, liquids, gases | Often requires dissolution; limited solid-state applications |
| Structural Specificity | Functional group level | Atomic level |
| Isotopic Discrimination | Limited | Excellent (e.g., C-H vs. C-D bonds) [96] |
| Detection Limits | Micrograms | Nanograms to micrograms |
| Complementary Techniques | Raman, NIR [97] [8] | MS, X-ray crystallography [93] |
Table 2: Functional Group Analysis Performance Comparison
| Functional Group | FTIR Detection Strength | NMR Detection Strength | Optimal Technique |
|---|---|---|---|
| Carbonyl (C=O) | Strong, distinct peak at 1700-1750 cm⁻¹ [14] | Indirect through neighboring protons | FTIR for initial screening |
| Hydroxyl (O-H) | Broad band at 3200-3600 cm⁻¹ [14] | Variable, concentration-dependent chemical shift | FTIR for hydrogen bonding studies |
| Aromatic | Multiple peaks in 1600-1500 cm⁻¹ region [14] | Distinct aromatic proton patterns (6.5-8.5 ppm) | Complementary |
| Alkene | C=C stretch at 1600-1680 cm⁻¹ [14] | Vinyl protons (4.5-6.5 ppm) | Complementary |
| Amine (N-H) | Sharp to broad peaks at 3300-3500 cm⁻¹ [14] | Broad, exchangeable protons | FTIR for primary detection |
| Nitrile (C≡N) | Sharp peak at ~2200 cm⁻¹ [14] | No direct proton signal | FTIR |
| Methyl (CH₃) | C-H stretch at ~2970, 2870 cm⁻¹ [14] | Distinct singlet or doublet (~0.7-1.3 ppm) | NMR for environment analysis |
Recent research demonstrates that a combined approach leveraging both techniques significantly enhances analytical accuracy. A 2025 machine learning study reported that an artificial neural network model trained on combined FTIR, ^1H NMR, and ^13C NMR data identified 17 functional groups with a macro-average F1 score of 0.93, outperforming models using any single technique alone (FTIR alone achieved 0.88) [5]. This synergistic effect is particularly valuable for analyzing functional groups with weak spectroscopic signals, such as nitriles, alkyl halides, and ethers.
Decision Matrix: Strategic Selection for Analytical Scenarios
Technique Selection Framework
The choice between FTIR and NMR depends on multiple factors including analytical objectives, sample characteristics, and resource constraints. The following decision matrix provides guidance for technique selection based on common analytical scenarios in functional group research:
Application-Specific Recommendations
Pharmaceutical Quality Control and Raw Material Testing: FTIR is ideally suited for rapid identity confirmation of raw materials and finished products due to its minimal sample preparation requirements, high throughput, and excellent fingerprinting capabilities [96]. Its non-destructive nature allows samples to be recovered for further testing. NMR becomes essential when investigating structural anomalies, elucidating impurity structures, or quantifying isotopic impurities as demonstrated in pharmaceutical case studies where FTIR quantified d0-, d1-, and d2-methylamine impurities down to ~0.3% in deuterated drugs [96].
Functional Group Identification in Synthetic Chemistry: FTIR provides rapid feedback during reaction monitoring, particularly for carbonyl formation, oxidation reactions, or hydroxyl consumption [8] [14]. However, NMR is indispensable for determining reaction regioselectivity, stereochemistry, and complete structural characterization of novel compounds. For complex functional groups with weak IR signals (nitriles, ethers), the combined approach proves most effective [5].
Polymer and Material Science: FTIR excels in characterizing polymer functional groups, degradation products, and surface modifications [92] [8]. ATR-FTIR accessories facilitate analysis of solid materials with minimal preparation. NMR provides deeper insights into copolymer composition, tacticity, and end-group analysis, though may require specialized solid-state NMR for insoluble materials.
Microplastics and Environmental Analysis: Recent research demonstrates the power of combining both techniques: FTIR identifies polymer types and oxidation (through carbonyl indices), while qNMR enables precise quantification of degradation products, with studies showing relative quantification errors of only 1-18% for aged microplastics [94].
Metabolomics and Complex Mixtures: NMR provides superior quantitative analysis without calibration and can identify unknown compounds in complex mixtures through 2D techniques [97] [93]. FTIR serves as a rapid screening tool for gross compositional changes but offers limited resolution for complex mixtures.
Experimental Protocols for Functional Group Analysis
FTIR Spectroscopy Protocol for Functional Group Identification
Methodology: This protocol describes the analysis of solid organic compounds using Attenuated Total Reflectance (ATR)-FTIR spectroscopy, adapted from recent pharmaceutical and materials research [5] [96] [14].
Materials and Equipment:
- FTIR spectrometer with ATR accessory (e.g., Specac Quest ATR or Harrick ConcentratIR2)
- Solid sample of interest
- Reference compounds for validation (optional)
- Methanol and lint-free wipes for cleaning
- Mortar and pestle for particle size reduction (if needed)
Step-by-Step Procedure:
Instrument Preparation:
- Power on the FTIR spectrometer and allow it to warm up for at least 15 minutes.
- Clean the ATR crystal thoroughly with methanol and lint-free wipes until no residue spectra are detected.
- Establish background spectrum with clean ATR crystal before sample analysis.
Sample Preparation:
- For solid samples, reduce particle size using a mortar and pestle to ensure good contact with the ATR crystal.
- Apply minimal sample quantity to cover the ATR crystal surface completely.
- Use consistent pressure application to ensure reproducible contact between sample and crystal.
Data Acquisition:
- Set scanning parameters to 4000-400 cm⁻¹ range with 4 cm⁻¹ resolution.
- Accumulate 32 scans per spectrum to enhance signal-to-noise ratio.
- Record spectra in absorbance mode.
- Maintain consistent atmospheric conditions (purge with dry air if necessary to minimize water vapor interference).
Spectral Processing:
Interpretation:
- Identify key absorption bands following the characteristic regions outlined in Table 2.
- Compare with reference spectra from databases for compound verification.
- Analyze peak shapes: broad O-H/N-H peaks indicate hydrogen bonding; sharp C≡N peaks suggest isolated polar bonds.
Troubleshooting:
- If spectra show excessive noise, increase scan number or check instrument alignment.
- If interference patterns appear, ensure sample has uniform contact with ATR crystal.
- If water vapor peaks obscure sample signals (3400 cm⁻¹ and 2300 cm⁻¹), extend purging time or check instrument seals [14].
NMR Spectroscopy Protocol for Structural Elucidation
Methodology: This protocol describes the acquisition and interpretation of ^1H and ^13C NMR spectra for organic compound structure verification, based on pharmaceutical analysis methods [5] [93].
Materials and Equipment:
- NMR spectrometer (e.g., Bruker AVANCE series, Jeol models)
- Deuterated solvent (CDCl₃ for general organic compounds, DMSO-d6 for polar compounds)
- NMR tubes (5 mm diameter)
- Reference compound (Tetramethylsilane, TMS)
- Sample of interest (1-10 mg depending on sensitivity)
Step-by-Step Procedure:
Sample Preparation:
- Dissolve 1-10 mg of sample in 0.6 mL of deuterated solvent.
- Add 1-2 drops of TMS reference standard (0.05% v/v) for chemical shift calibration.
- Transfer solution to a clean, dry NMR tube using a Pasteur pipette.
Instrument Setup:
- Insert sample tube into spectrometer and allow temperature equilibration.
- Lock instrument on deuterium signal of the solvent.
- Shim magnet to optimize field homogeneity.
- Set probe temperature to 25°C (unless temperature studies are required).
^1H NMR Acquisition:
- Set spectral width to 12-15 ppm.
- Set pulse program to standard single-pulse experiment (zg30).
- Set relaxation delay (D1) to 1-5 seconds based on estimated T1.
- Acquire 16-64 scans with 64k data points.
- Apply exponential line broadening (0.3 Hz) before Fourier transformation.
^13C NMR Acquisition:
- Set spectral width to 220-240 ppm.
- Use inverse-gated decoupling to minimize Nuclear Overhauser Effect for quantitative analysis.
- Acquire 128-1024 scans with 128k data points depending on sample concentration.
- Set relaxation delay to 2-5 seconds (longer for quantitative experiments).
Data Processing:
- Apply Fourier transformation to time-domain data.
- Phase correct spectra manually for optimal baseline.
- Reference spectra to TMS signal (0.0 ppm).
- Integrate ^1H NMR signals for quantitative analysis.
Interpretation:
- Analyze chemical shifts, integration values, and coupling patterns in ^1H NMR.
- Correlate ^13C chemical shifts with expected functional groups.
- For complex structures, employ 2D techniques (COSY, HSQC, HMBC) to establish connectivity.
Troubleshooting:
- If signal-to-noise is poor, increase scan number or concentration.
- If resolution is inadequate, check shimming or reduce sample volume.
- If solvent peaks are too intense, consider using different deuterated solvent.
Integrated Workflows and Advanced Applications
Multi-Technique Approaches for Complex Analyses
Research demonstrates that the most powerful analytical strategies often combine FTIR and NMR in complementary workflows. A 2025 study on microplastic analysis exemplifies this approach, where FTIR initially identified polymer types and surface oxidation through carbonyl indices, while qNMR provided precise quantification of degradation products with detection limits between 0.87-2.79 μg mL⁻¹ [94]. This integrated methodology delivered comprehensive insights into aging processes that neither technique could provide independently.
In pharmaceutical development, Bruker advocates for a "wide range of techniques" approach where FTIR provides rapid functional group information, while NMR and MS data establish molecular connectivity and fragment assembly [93]. This workflow is particularly valuable for impurity profiling, where thresholds as low as 0.1% require both sensitive detection (FTIR) and definitive structural elucidation (NMR) [93].
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Essential Materials for FTIR and NMR Analysis
| Item | Function | Application Notes |
|---|---|---|
| ATR-FTIR Accessory (e.g., Specac Quest, Harrick ConcentratIR2) | Enables solid and liquid sample analysis with minimal preparation | Multi-bounce crystals enhance sensitivity for trace analysis [96] |
| Deuterated Solvents (CDCl₃, DMSO-d₆) | Provides locking signal for NMR; dissolves analytes | Choice affects chemical shifts; DMSO-d₆ for polar compounds, CDCl₃ for general organics |
| NMR Reference Standards (TMS, DSS) | Chemical shift calibration | TMS (0.0 ppm) for organic solvents; DSS for aqueous solutions |
| FTIR Spectral Databases | Reference for compound identification | Commercial and custom libraries for fingerprint matching |
| qNMR Standards (maleic acid, DMSO) | Quantitative calibration for NMR | Certified reference materials with known purity [95] |
| Chemometric Software (PCA, PLS algorithms) | Extracts meaningful information from complex spectral data | Essential for quantitative FTIR and mixture analysis [5] [96] |
FTIR and NMR spectroscopy represent complementary rather than competing technologies in the analytical scientist's arsenal. FTIR excels in rapid functional group identification, particularly for carbonyl, hydroxyl, and unsaturated compounds, with minimal sample preparation and high throughput capabilities. NMR provides unparalleled structural elucidation power, quantitative accuracy, and atomic-level insight into molecular architecture. The strategic selection between these techniques should be guided by analytical objectives, sample characteristics, and required information level, as outlined in the decision matrix.
Current research trends indicate growing emphasis on integrated approaches, where machine learning models combine data from multiple spectroscopic techniques to achieve superior accuracy in functional group identification [5]. Furthermore, advancements in portability (for FTIR) and automation (for NMR) are expanding application possibilities across field analysis and high-throughput screening environments. For researchers in drug development and functional group analysis, mastering both techniques and understanding their synergistic relationship remains essential for solving complex structural challenges efficiently and accurately.
In the structural elucidation of unknown compounds, particularly in natural product discovery and drug development, reliance on a single analytical technique introduces significant risk of misidentification. While Fourier-Transform Infrared (FTIR) and Nuclear Magnetic Resonance (NMR) spectroscopy are foundational techniques for functional group identification and structural analysis, they present inherent limitations when used in isolation [12] [1]. FTIR spectroscopy provides excellent information on vibrational and rotational modes of chemical bonds, making it ideal for identifying functional groups. NMR spectroscopy, conversely, offers detailed insights into the nuclear environment, molecular connectivity, and stereochemistry [1]. However, each method provides only partial information about a molecule, and spectral interpretation can be prone to error, often relying on the intuition and experience of the analyst [5].
The integration of Mass Spectrometry (MS) with FTIR and NMR creates a powerful synergistic workflow, establishing a "gold standard" for validation. This cross-corroborative approach significantly enhances the confidence, accuracy, and comprehensiveness of structural assignments [98]. MS contributes critical data on molecular weight and fragment patterns, providing a different dimension of structural information that complements and confirms findings from spectroscopic techniques. This multi-technique strategy is essential for overcoming the limitations of individual methods, ultimately leading to more reliable characterization of complex molecules in research and development.
Comparative Analysis of Core Analytical Techniques
A thorough understanding of the strengths and limitations of each analytical technique is a prerequisite for designing an effective validation strategy. The following table provides a structured comparison of FTIR, NMR, and Mass Spectrometry.
Table 1: Comparison of FTIR, NMR, and Mass Spectrometry for Structural Analysis
| Feature | FTIR Spectroscopy | NMR Spectroscopy | Mass Spectrometry (MS) |
|---|---|---|---|
| Primary Information | Vibrational/rotational modes of bonds; functional groups [12] [1] | Nuclear spin transitions; molecular connectivity, stereochemistry, dynamics [1] | Mass-to-charge ratio (m/z) of ions; molecular mass, fragment patterns [98] |
| Key Strengths | Rapid functional group identification; simple sample prep; versatile sample types [12] [84] | Detailed atomic-level structural and dynamic information; quantitative capability [98] | High sensitivity and resolution; provides molecular formula data; detects trace components [98] |
| Common Limitations | Limited to identifying group types, not full structure; peak overlap in complex mixtures [12] | Lower sensitivity requires higher concentration; complex data analysis; expensive [98] | Destructive to sample; requires ionization, which can cause fragmentation; no direct stereochemical info [98] |
| Complementary Role in Validation | Provides initial functional group screening and hypothesis generation. | Confirms molecular skeleton, connectivity, and functional group environment. | Confirms molecular weight, provides elemental composition, and supports structural hypotheses via fragments. |
The synergy between these techniques is profound. FTIR offers a rapid initial profile, NMR provides atomic-resolution blueprint of the carbon skeleton and its substituents, and MS delivers precise molecular mass and "fingerprint" fragments. For instance, an FTIR signal suggesting a carbonyl group (C=O stretch around 1700 cm⁻¹) can be ambiguous, but when combined with NMR, it can be definitively assigned as an aldehyde, ketone, or amide. MS can then confirm the assignment by revealing a mass consistent with the proposed structure or showing a characteristic fragment loss [12] [1] [98].
Experimental Protocols for Cross-Corroborative Analysis
Protocol 1: Integrated Workflow for Functional Group Identification and Validation
This protocol outlines a systematic approach for analyzing an unknown organic compound by integrating data from FTIR, NMR, and MS.
I. Sample Preparation
- FTIR: For liquids, use a drop between two KBr plates. For solids, prepare a KBr pellet or use an Attenuated Total Reflectance (ATR) accessory with minimal sample preparation [84].
- NMR: Dissolve 2-10 mg of sample in 0.5-0.7 mL of a deuterated solvent (e.g., CDCl₃, DMSO-d₆). Filter the solution if particulate matter is present.
- MS: Prepare a dilute solution (ppm to ppb range) in a volatile solvent compatible with the ionization technique (e.g., methanol, acetonitrile).
II. Data Acquisition
- FTIR Analysis:
- NMR Analysis:
- Acquire ¹H NMR and ¹³C NMR spectra.
- For complex molecules, acquire 2D experiments (e.g., COSY, HSQC, HMBC) to establish connectivity [1].
- Note chemical shifts (δ, ppm), integration (for ¹H), and multiplicity (for ¹H).
- MS Analysis:
- Acquire data using an appropriate ionization method (e.g., EI, ESI, APCI).
- Obtain a high-resolution mass spectrum to determine the exact mass and propose a molecular formula.
- Analyze fragmentation patterns in the MS/MS spectrum for structural clues.
III. Data Integration and Validation
- Hypothesis Generation: Use the FTIR spectrum to generate an initial list of potential functional groups present in the molecule.
- Structural Elucidation: Use the NMR data (chemical shifts, coupling, and 2D correlations) to build potential carbon skeletons and assign the functional groups identified by FTIR to specific positions within the molecule.
- Cross-Corroboration with MS:
- Confirm the molecular formula proposed from the NMR data with the exact mass from MS.
- Verify that key fragmentation patterns in the MS data are consistent with the proposed structure. For example, the loss of a specific group (e.g., -OH, -OCH₃) should be explainable by the structure built from NMR data.
- Consistency Check: Ensure all data are internally consistent. Any discrepancy must be investigated, as it may indicate an error in interpretation or a more complex structural feature.
Diagram 1: Integrated functional group analysis workflow.
Protocol 2: Machine Learning-Enhanced Functional Group Prediction
Recent advances demonstrate the power of machine learning (ML) to automate and enhance the cross-corroboration process. This protocol is based on a study that used an Artificial Neural Network (ANN) to identify 17 functional groups by simultaneously training on FTIR, ¹H NMR, and ¹³C NMR data [5].
I. Data Collection and Preprocessing
- Data Sourcing: Collect FTIR, ¹H NMR, and ¹³C NMR spectra for a large dataset of compounds (e.g., 3027 compounds) from reference databases like NIST Chemistry WebBook and SDBS [5].
- FTIR Processing: Transform spectra into vectors (e.g., 400-4000 cm⁻¹). Convert transmittance to absorbance and apply min-max normalization.
- NMR Processing (Data Binning):
- To reduce data dimensionality and sparsity, use a binning approach.
- For ¹H NMR, divide the 1-12 ppm range into bins (e.g., 12 bins with a 1 ppm interval).
- For ¹³C NMR, divide the 1-220 ppm range into bins (e.g., 44 bins with a 5 ppm interval).
- Assign a value of 1 or 0 to each bin to indicate the presence or absence of a peak, ignoring intensity [5].
- Functional Group Labeling: Use SMARTS strings to algorithmically assign the presence or absence of specific functional groups for each compound in the dataset, creating the target data for the model [5].
II. Model Training and Validation
- Model Selection: Implement an Artificial Neural Network (ANN) with an input layer, multiple hidden layers, and an output layer corresponding to the functional groups.
- Stratified K-Fold Cross-Validation: Partition the data into training and testing sets (e.g., 80/20 split). Use multilabel stratified 5-fold cross-validation on the training data to avoid overfitting and create a generalized model [5] [99].
- Performance Evaluation: Evaluate the model using metrics such as the F1 score. The integrated model (using all three spectral types) has been shown to outperform models using any single spectroscopic technique (macro-average F1 score of 0.93 vs. 0.88 for FTIR alone) [5].
III. Application to Unknowns
- Data Input: Preprocess the FTIR and NMR spectra of an unknown compound using the same binning and normalization procedures as in the training phase.
- Prediction: The trained ANN model will output a probability for the presence of each of the functional groups it was trained on.
- Expert Review: The ML-generated functional group profile serves as a highly accurate starting point for the scientist, who can then perform a more targeted and efficient structural elucidation.
Table 2: Key Research Reagents and Materials for Cross-Corroborative Analysis
| Category | Item | Function/Description |
|---|---|---|
| Solvents | Deuterated Solvents (CDCl₃, DMSO-d₆) | Provides the lock signal for NMR spectrometers and dissolves samples without adding interfering proton signals [5]. |
| HPLC/MS Grade Volatile Solvents (MeOH, ACN) | High-purity solvents for MS analysis to minimize background noise and adduct formation. | |
| Sample Preparation | Potassium Bromide (KBr) | Used for preparing pellets for FTIR transmission analysis of solid samples [84]. |
| ATR Crystals (Diamond, ZnSe) | Durable crystals in ATR-FTIR accessories that allow for direct analysis of solids and liquids with minimal preparation [84]. | |
| Data Analysis | Spectral Databases (NIST, SDBS, BMRB) | Reference libraries for comparing acquired FTIR, MS, and NMR spectra to identify known compounds [5] [98]. |
| SMARTS Strings | A line notation used to encode molecular structures and algorithmically assign functional groups for machine learning applications [5]. |
Case Study: Metabolite Identification in Complex Biological Mixtures
A compelling illustration of this gold-standard approach comes from a metabolomics study on Chlamydomonas reinhardtii [98]. The research aimed to characterize metabolic changes induced by chemical treatments and underscores the complementarity of NMR and MS.
The study design involved analyzing aqueous extracts of treated and untreated cells using both 1D/2D NMR and GC-MS. The results were striking:
- NMR uniquely identified 14 metabolites of interest, including glycine, lysine, and key TCA cycle intermediates like succinate and malate.
- GC-MS uniquely identified 16 metabolites, such as asparagine, serine, and fructose-6-phosphate.
- Both techniques identified 17 common metabolites, including several nucleotides like adenosine and guanosine [98].
This distribution clearly demonstrates that reliance on a single technique would have missed a significant portion of the metabolome, leading to an incomplete and potentially misleading biological interpretation. The combined NMR-MS approach provided a more comprehensive coverage of central metabolic pathways, including the oxidative pentose phosphate pathway, Calvin cycle, and amino acid biosynthesis. Furthermore, for the metabolites identified by both techniques, the results showed consistent changes upon treatment, thereby cross-validating the findings and increasing the overall confidence in the conclusions [98].
Diagram 2: Metabolite identification synergy in a case study.
The integration of FTIR, NMR, and Mass Spectrometry represents an unequivocal gold standard for the validation of molecular structures in functional group research and beyond. This cross-corroborative framework effectively mitigates the limitations intrinsic to any single analytical method, transforming structural elucidation from a speculative exercise into a robust, data-driven process. The synergy between these techniques—where FTIR provides functional group clues, NMR constructs the molecular skeleton, and MS confirms molecular mass and formula—creates a powerful cycle of hypothesis generation and testing.
The adoption of this multi-technique paradigm, now further enhanced by machine learning models that integrate multi-spectral data, is critical for advancing research in complex fields such as natural product chemistry, drug development, and metabolomics [100] [5]. By deliberately moving beyond instrument-specific silos, researchers can achieve unparalleled accuracy, depth, and confidence in their analytical findings, ensuring that the identification of a molecular structure is not just a conclusion, but a thoroughly validated scientific fact.
The structural elucidation of complex molecules, such as those found in pharmaceuticals, natural products, and synthetic polymers, is a cornerstone of modern chemical research and drug development. While numerous analytical techniques exist, Fourier Transform Infrared (FTIR) spectroscopy and High-Resolution Nuclear Magnetic Resonance (HR-NMR) spectroscopy stand out for their ability to provide complementary molecular information. FTIR spectroscopy is a powerful technique for identifying functional groups based on their characteristic vibrational frequencies when exposed to infrared light [92]. Concurrently, HR-NMR spectroscopy offers unparalleled insights into the molecular backbone, detailing the carbon-hydrogen framework and the connectivity between atoms within a molecule [101]. This application note details a structured protocol for the integrated use of FTIR and NMR to achieve comprehensive molecular identification, framed within a broader thesis on qualitative analysis techniques for functional group research. The synergistic application of these methods is particularly powerful for analyzing complex mixtures and confirming the identity of novel compounds, providing a robust framework for researchers and drug development professionals.
Theoretical Foundations of FTIR and NMR
Fourier Transform Infrared (FTIR) Spectroscopy
FTIR spectroscopy operates on the principle that chemical bonds within a molecule vibrate at specific frequencies when irradiated with infrared light [92]. These vibrations, which include stretching and bending modes, are unique to different functional groups. An FTIR instrument works by passing a broad spectrum of infrared light through an interferometer, which creates an interference pattern (interferogram) after the light interacts with the sample. A mathematical operation known as a Fourier transform is then applied to this raw data, converting it into a recognizable absorption spectrum that plots absorbance against wavenumber (cm⁻¹) [92]. Key spectral regions include:
- 1700 cm⁻¹: Often indicates a carbonyl (C=O) group.
- 3200–3600 cm⁻¹: A broad peak suggesting hydroxyl (O–H) groups [92]. This "molecular fingerprint" allows for the rapid identification of functional groups present in a sample.
High-Resolution Nuclear Magnetic Resonance (NMR) Spectroscopy
HR-NMR spectroscopy is a non-invasive analytical technique that exploits the magnetic properties of certain atomic nuclei, such as ¹H and ¹³C. When placed in a strong magnetic field, these nuclei absorb and re-emit electromagnetic radiation at frequencies characteristic of their chemical environment [101]. This provides detailed information on:
- Molecular structure and dynamics: Revealing the atomic connectivity within a molecule.
- Composition and purity: Allowing for the identification and quantification of components in a mixture [102]. Advanced NMR techniques, such as multidimensional NMR (e.g., COSY, HSQC), are essential for resolving complex spectra with overlapping signals, thereby providing unambiguous structural assignment for complex molecules [101].
Experimental Protocol for Integrated Analysis
Sample Preparation
- FTIR Analysis: For solid samples, prepare a homogeneous pellet by grinding 1-2 mg of the sample with 100-200 mg of potassium bromide (KBr). For liquid samples, a drop can be sandwiched between sodium chloride (NaCl) plates. Ensure samples are free of moisture to avoid spectral interference from water.
- NMR Analysis: Dissolve 2-10 mg of the sample in 0.6-0.7 mL of a suitable deuterated solvent (e.g., CDCl₃, DMSO-d₆). Transfer the solution to a clean, dry NMR tube for analysis. For quantitative NMR (qNMR), use an internal standard such as tetramethylsilane (TMS) or a known quantity of a maleic acid for precise concentration determination [101] [102].
Instrumentation and Data Acquisition
- FTIR Instrument Settings: Use a spectrometer with a resolution of 4 cm⁻¹. Accumulate 32 scans per spectrum to ensure a high signal-to-noise ratio. The measurement range should typically be between 4000 and 400 cm⁻¹.
- NMR Data Acquisition: Conduct ¹H NMR analysis at a field strength of 400 MHz or higher for sufficient resolution. For complex mixtures, employ two-dimensional techniques such as ¹H-¹³C Heteronuclear Single Quantum Coherence (HSQC) to correlate proton and carbon signals and Diffusion-Ordered Spectroscopy (DOSY) to separate components by their molecular size [101] [102]. For samples with significant signal overlap, pure-shift NMR methods like PSYCHE can be applied to achieve broadband homonuclear decoupling and simplify the spectrum [102].
Data Interpretation and Integration
- Begin with FTIR Analysis: Inspect the FTIR spectrum to identify major functional groups present (e.g., OH, C=O, aromatic C-H).
- Proceed to ¹H NMR: Use the chemical shift (δ, ppm), integration, and spin-spin coupling (J, Hz) patterns from the ¹H NMR spectrum to identify the types and numbers of protons and their neighboring atoms.
- Employ ¹³C NMR and 2D Techniques: Use the ¹³C NMR spectrum and 2D correlations (e.g., HSQC, COSY) to map out the carbon framework and connect the proton-bearing fragments.
- Integrate Findings: Correlate the functional groups identified by FTIR with the specific structural environments and connectivities revealed by NMR to propose a complete molecular structure. For instance, a carbonyl peak at 1700 cm⁻¹ in FTIR could be assigned to a specific ketone, aldehyde, or amide group based on its chemical shift in the ¹³C NMR spectrum and its correlations in an HSQC experiment.
Research Reagent Solutions
The following table details essential materials and their functions for the experiments described in this protocol.
Table 1: Essential Research Reagents and Materials
| Item | Function/Brief Explanation |
|---|---|
| Deuterated Solvents (e.g., CDCl₃, DMSO-d₆) | Provides a non-interfering, deuterium-based medium for NMR analysis, enabling the instrument's lock system to function. |
| Potassium Bromide (KBr) | An IR-transparent salt used to form pellets for solid sample analysis in FTIR spectroscopy. |
| Internal Standard (e.g., TMS) | A reference compound added in qNMR with a known concentration and a defined chemical shift (δ 0.00 ppm) for precise quantification of sample components [102]. |
| NMR Tube | A high-precision, thin-walled glass tube designed to hold the sample within the NMR spectrometer's magnetic field. |
| Hydrofluoric Acid (HF) | Used in pre-treatment of solid samples (e.g., soils) to remove silicate minerals, thereby reducing interference and improving the accuracy of FTIR analysis for organic components [17]. |
Workflow and Signaling Pathways
The following diagram illustrates the logical workflow for the integrated FTIR-NMR analysis, from sample preparation to final identification.
Integrated FTIR-NMR Analysis Workflow
Comparative Data Presentation
The table below summarizes the key quantitative and qualitative aspects of FTIR and NMR spectroscopy for easy comparison, highlighting their complementary roles.
Table 2: Comparative Analysis of FTIR and NMR Techniques
| Parameter | FTIR Spectroscopy | NMR Spectroscopy |
|---|---|---|
| Primary Information | Functional group identification based on molecular bond vibrations [92]. | Molecular structure, dynamics, and atomic connectivity based on nuclear spin transitions [101]. |
| Sample Form | Solids (KBr pellets), liquids, gases [92]. | Primarily liquids (solutions), solids require specialized techniques (ssNMR) [101]. |
| Key Spectral Features | Absorption bands at specific wavenumbers (e.g., 1700 cm⁻¹ for C=O) [92]. | Chemical shift (δ), integration, coupling constants (J) [101]. |
| Advantages in Mixture Analysis | Rapid screening for functional groups; can be enhanced by HF pretreatment to remove mineral interference [17]. | Powerful for resolving complex mixtures using techniques like DOSY and pure-shift methods [102]. |
| Quantitative Capability | Possible, but can be less straightforward due to baseline issues. | Excellent via Quantitative NMR (qNMR); peak area is directly proportional to the number of nuclei [101] [102]. |
| Complementary Role | Provides a initial "functional group map" of the sample. | Validates and provides detailed context for the functional groups identified by FTIR, placing them within the full molecular structure. |
Application Notes
The integration of multi-spectral data represents a paradigm shift in analytical chemistry, particularly for the identification of functional groups—a fundamental task in elucidating the structure and properties of unknown compounds [5]. Traditional analysis, which relies on the sequential interpretation of individual spectra (e.g., FTIR, ¹H NMR, ¹³C NMR), is often time-consuming and susceptible to human error due to its dependence on analyst intuition and experience [5]. The "hybrid" approach leverages machine learning (ML) models, specifically artificial neural networks (ANNs), trained simultaneously on multiple types of spectral data. This mirrors the expert practice of cross-referencing spectra but does so with superior speed and accuracy, enabling more reliable functional group prediction and facilitating advances in drug development and materials science [5].
Key Performance Findings
Quantitative analysis demonstrates that models trained on fused multi-spectral data significantly outperform those using a single spectral type. The following table summarizes the performance of an ANN model in identifying 17 key functional groups.
Table 1: Performance of ANN Model in Functional Group Identification [5]
| Functional Group | Spectral Data Combination | Macro-Average F1 Score |
|---|---|---|
| Aromatic, Ether, Alcohol, Ester, Methyl, Nitro, Alkane, Carboxylic Acid, Amine, Aldehyde, Alkyne, Ketone, Alkyl Halide, Amide, Alkene, Nitrile, Acyl Halide | FT-IR + ¹H NMR + ¹³C NMR | 0.93 |
| Same 17 Functional Groups | FT-IR only | 0.88 |
This performance improvement is crucial for identifying challenging functional groups like nitriles, alkyl halides, and ethers, which often produce weak signals in individual spectra [5]. The hybrid model's ability to learn from complementary data sources makes it a powerful tool for researchers.
Experimental Protocols
Protocol 1: Data Collection and Preprocessing for Multi-Spectral ML
Purpose: To gather and standardize FTIR and NMR spectral data for training a machine learning model.
Materials:
- Data Sources: NIST Chemistry WebBook (for FT-IR spectra) and the Spectral Database for Organic Compounds (SDBS) (for ¹H and ¹³C NMR spectra) [5].
- Software: Standard data processing libraries (e.g., Python with Pandas, NumPy).
Methodology:
- Data Collection:
- FT-IR Data Preprocessing:
- Transform spectra into a vector representing wavelengths from 400 to 4000 cm⁻¹.
- Convert transmittance values to absorbance.
- Apply min-max normalization by dividing each absorbance value by the maximum value in the dataset.
- Estimate any missing values using linear interpolation [5].
- NMR Data Preprocessing (Data Binning):
- To reduce data dimensionality and sparsity, employ a binning approach.
- For ¹H NMR, divide the 1–12 ppm range into 12 bins, each with a 1 ppm interval.
- For ¹³C NMR, divide the 1–220 ppm range into 44 bins, each with a 5 ppm interval.
- For each bin, assign a value of
1if a peak is present and0if absent. Ignore intensity information for this model [5].
- Functional Group Labeling:
- Use SMARTS strings to programmatically assign the presence (1) or absence (0) of 17 target functional groups for each compound, creating the target labels for the ML model [5].
Protocol 2: Building and Training the Artificial Neural Network
Purpose: To construct and evaluate an ML model for functional group identification from preprocessed multi-spectral data.
Materials:
- Computing Environment: Python with deep learning frameworks such as TensorFlow or PyTorch.
- Hardware: Computer with a CUDA-enabled GPU for accelerated training (recommended).
Methodology:
- Model Architecture:
- Design an Artificial Neural Network (ANN) with an input layer, multiple hidden layers, and an output layer.
- The input layer must accommodate the combined dimensions of the preprocessed FT-IR, ¹H NMR, and ¹³C NMR vectors.
- The output layer should have 17 nodes, each corresponding to one of the functional groups, using a sigmoid activation function for multi-label classification [5].
- Model Training and Validation:
- Partition the dataset, using 20% as a hold-out test set.
- On the remaining 80%, perform Multilabel Stratified 5-Fold Cross-Validation:
- Split the data into 5 subsets.
- Iteratively use 4 subsets for training and 1 for validation.
- Repeat this process 5 times, ensuring each subset serves as the validation set once [5].
- This rigorous method prevents overfitting and leads to a more generalized model.
- Performance Evaluation:
- Evaluate the final model's performance on the held-out test set using the macro-average F1 score across all 17 functional groups [5].
Workflow and System Diagrams
Simplified Multi-Spectral ML Workflow
The following diagram illustrates the end-to-end process for building a hybrid machine learning model for functional group identification.
Data Preprocessing Pipeline
This diagram details the critical data preprocessing steps for preparing spectral data for the machine learning model.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials and Data Sources for Multi-Spectral ML Experiments
| Item | Function / Purpose | Specification / Note |
|---|---|---|
| NIST Chemistry WebBook | Source for experimental FT-IR spectral data. | Provides gas-phase spectra; crucial for building a standardized training dataset [5]. |
| SDBS Database | Source for experimental ¹H and ¹³C NMR spectral data. | Ensure spectral consistency by selecting data obtained in a common solvent like CDCl₃ [5]. |
| CDCl₃ Solvent | Standard solvent for NMR spectroscopy. | Using a single, common solvent for all NMR data minimizes chemical shift variability, a key pre-processing step [5]. |
| SMARTS Strings | Line notation for encoding molecular structures. | Used to programmatically assign functional group labels to each compound in the dataset, creating ground-truth for model training [5]. |
| Artificial Neural Network (ANN) | Core machine learning algorithm. | Capable of learning complex, non-linear relationships from the high-dimensional, multi-spectral input data [5]. |
| Stratified K-Fold Cross-Validation | Model validation technique. | Ensures robust model performance estimation and prevents overfitting by maintaining the distribution of all functional groups across all data splits [5]. |
Conclusion
FTIR and NMR spectroscopy are not competing but profoundly complementary techniques that form the bedrock of functional group analysis in modern research. FTIR excels in rapid functional group screening and providing insights into molecular symmetry and bonding, while NMR offers unparalleled detail on atomic connectivity and three-dimensional molecular structure. The integration of these methods, especially when enhanced by machine learning and chemometrics, provides a robust framework for the accurate identification and validation of chemical structures. For biomedical and clinical research, future directions point toward the increased use of portable FTIR devices for point-of-care diagnostics, the application of integrated spectroscopic models for high-throughput drug screening, and the deeper exploration of protein-ligand interactions and lipidomics. Embracing these synergistic and technologically advanced approaches will accelerate discovery and innovation in drug development and clinical diagnostics.
References
- 1. Difference between FTIR and NMR? - Rocky Mountain Labs [https://rockymountainlabs.com/difference-between-ftir-and-nmr/]
- 2. Applications of Single-Molecule Vibrational Spectroscopic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9573641/]
- 3. An FTIR, GC–MSD, and NMR Study [https://www.mdpi.com/1420-3049/30/14/2927]
- 4. Leveraging infrared spectroscopy for automated structure ... [https://www.nature.com/articles/s42004-024-01341-w]
- 5. Machine-Learning Approach to Identify Organic Functional ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11966250/]
- 6. FTIR vs NMR: Comprehensive Molecular Structure Insights [https://eureka.patsnap.com/report-ftir-vs-nmr-comprehensive-molecular-structure-insights]
- 7. Trace element detection in anhydrous minerals by micro ... [https://www.nature.com/articles/s41467-024-51131-0]
- 8. A Review of the Latest Research Applications Using FT-IR ... [https://www.spectroscopyonline.com/view/a-review-of-the-latest-research-applications-using-ft-ir-spectroscopy]
- 9. Fourier-transform infrared spectroscopy [https://en.wikipedia.org/wiki/Fourier-transform_infrared_spectroscopy]
- 10. FTIR Analysis Beginner's Guide: Interpreting Results [https://www.innovatechlabs.com/newsroom/1882/interpreting-analyzing-ftir-results/]
- 11. Guide to FT-IR Spectroscopy [https://www.bruker.com/en/products-and-solutions/infrared-and-raman/ft-ir-routine-spectrometer/what-is-ft-ir-spectroscopy.html]
- 12. IR Spectroscopy in Qualitative and Quantitative Analysis [https://www.intechopen.com/chapters/83478]
- 13. Infrared Spectral Interpretation, In The Beginning I [https://www.spectroscopyonline.com/view/infrared-spectral-interpretation-in-the-beginning-i-the-meaning-of-peak-positions-heights-and-widths]
- 14. How to Interpret FTIR Results: A Beginner's Guide [https://www.azooptics.com/Article.aspx?ArticleID=2756]
- 15. A Process for Successful Infrared Spectral Interpretation [https://www.spectroscopyonline.com/view/process-successful-infrared-spectral-interpretation]
- 16. FTIR Spectroscopy Resources [https://www.thermofisher.com/us/en/home/industrial/spectroscopy-elemental-isotope-analysis/molecular-spectroscopy/fourier-transform-infrared-spectroscopy/resources.html]
- 17. Article Comparison of infrared and solid-state 13 C NMR ... [https://www.sciencedirect.com/science/article/pii/S2667325824005326]
- 18. 19.4: Applications of Proton NMR [https://chem.libretexts.org/Bookshelves/Analytical_Chemistry/Instrumental_Analysis_(LibreTexts)/19%3A_Nuclear_Magnetic_Resonance_Spectroscopy/19.04%3A_Applications_of_Proton_NMR]
- 19. Proton NMR Table - MSU chemistry [https://www2.chemistry.msu.edu/faculty/reusch/orgpage/nmr.htm]
- 20. Benefits from nuclear magnetic resonance studies of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12124707/]
- 21. Applications of high-field nuclear magnetic resonance ... [https://www.sciencedirect.com/science/article/pii/S2666441025000159]
- 22. The use of FTIR and NMR spectroscopies to study ... [https://pubmed.ncbi.nlm.nih.gov/18246331/]
- 23. Capturing Local Compositional Fluctuations in NMR ... [https://chemrxiv.org/engage/chemrxiv/article-details/685556b93ba0887c338af008]
- 24. Capturing local compositional fluctuations in NMR ... [https://pubmed.ncbi.nlm.nih.gov/40988690/]
- 25. Applications of Fourier Transform-Infrared spectroscopy in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10703385/]
- 26. The Fingerprint Region [https://chem.libretexts.org/Bookshelves/Physical_and_Theoretical_Chemistry_Textbook_Maps/Supplemental_Modules_(Physical_and_Theoretical_Chemistry)/Spectroscopy/Vibrational_Spectroscopy/Infrared_Spectroscopy/The_Fingerprint_Region]
- 27. Infrared Spectroscopy [https://www2.chemistry.msu.edu/faculty/reusch/virttxtjml/spectrpy/infrared/infrared.htm]
- 28. 12.8: Infrared Spectra of Some Common Functional Groups [https://chem.libretexts.org/Bookshelves/Organic_Chemistry/Organic_Chemistry_(Morsch_et_al.)/12%3A_Structure_Determination_-_Mass_Spectrometry_and_Infrared_Spectroscopy/12.08%3A_Infrared_Spectra_of_Some_Common_Functional_Groups]
- 29. Interpreting Infrared Spectra [https://specac.com/theory-articles/interpreting-infra-red-spectroscopy/]
- 30. Infrared (IR) Spectroscopy: A Quick Primer On Interpreting ... [https://www.masterorganicchemistry.com/2016/11/23/quick_analysis_of_ir_spectra/]
- 31. FTIR Functional Group Database Table with Search [https://instanano.com/all/characterization/ftir/ftir-functional-group-search/]
- 32. 10.7: Functional Groups and IR Tables [https://chem.libretexts.org/Ancillary_Materials/Laboratory_Experiments/Wet_Lab_Experiments/Organic_Chemistry_Labs/Lab_I/10%3A_Infrared_Spectroscopy/10.07%3A_Functional_Groups_and_IR_Tables]
- 33. NMR Spectroscopy [https://www2.chemistry.msu.edu/faculty/reusch/virttxtjml/spectrpy/nmr/nmr1.htm]
- 34. The Basics of Interpreting a Proton ( 1 H) NMR Spectrum [https://www.acdlabs.com/blog/the-basics-of-interpreting-a-proton-nmr-spectrum/]
- 35. 4.7: NMR Spectroscopy [https://chem.libretexts.org/Bookshelves/Analytical_Chemistry/Physical_Methods_in_Chemistry_and_Nano_Science_(Barron)/04%3A_Chemical_Speciation/4.07%3A_NMR_Spectroscopy]
- 36. 13.3: Chemical Shifts in ¹H NMR Spectroscopy [https://chem.libretexts.org/Bookshelves/Organic_Chemistry/Organic_Chemistry_(Morsch_et_al.)/13%3A_Structure_Determination_-_Nuclear_Magnetic_Resonance_Spectroscopy/13.03%3A_Chemical_Shifts_in_H_NMR__Spectroscopy]
- 37. NMR - Interpretation [https://chem.libretexts.org/Bookshelves/Physical_and_Theoretical_Chemistry_Textbook_Maps/Supplemental_Modules_(Physical_and_Theoretical_Chemistry)/Spectroscopy/Magnetic_Resonance_Spectroscopies/Nuclear_Magnetic_Resonance/NMR%3A_Experimental/NMR_-_Interpretation]
- 38. 12.5: Functional Groups and Chemical Shifts in ¹H NMR ... [https://chem.libretexts.org/Bookshelves/Organic_Chemistry/Map%3A_Organic_Chemistry_(Wade)_Complete_and_Semesters_I_and_II/Map%3A_Organic_Chemistry_(Wade)/12%3A_Nuclear_Magnetic_Resonance_Spectroscopy/12.05%3A_Functional_Groups_and_Chemical_Shifts_in_H_NMR__Spectroscopy]
- 39. NMR Chemical Shift Values Table [https://www.chemistrysteps.com/nmr-chemical-shift-values-table/]
- 40. Analytical NMR [https://magritek.com/applications/analytical-nmr/]
- 41. nmrshiftdb2 - open nmr database on the web [https://nmrshiftdb.nmr.uni-koeln.de/]
- 42. CASCADE-2.0: Real Time Prediction of 13C-NMR Shifts ... [https://chemrxiv.org/engage/chemrxiv/article-details/68828a5a23be8e43d6d83671]
- 43. Sample Preparation - Free Micro ATR FT-IR Chemical ... [https://www.spectroscopyonline.com/view/sample-preparation-free-micro-atr-ft-ir-chemical-imaging-polymers-and-polymer-laminates]
- 44. KBr Pellet Press - UW Department of Chemistry [https://chem.washington.edu/instruments/kbr-pellet-press]
- 45. FT‐IR Sample Preparation - NIU - Department of Chemistry ... [https://www.niu.edu/chembio/research/analytical-lab/ftir/sample-preparation.shtml]
- 46. Fourier transform infrared spectroscopic technique for analysis ... [https://pubs.rsc.org/en/content/articlehtml/2025/na/d5na00522a]
- 47. Making KBr Pellets for FTIR: Step by Step Guide [https://www.pelletpressdiesets.com/pages/kbr-pellet-press]
- 48. Desirable Characteristics of Solvents used for NMR Studies [https://lab-training.com/desirable-characteristics-of-solvents-used-for-nmr-studies/]
- 49. the powerful combination of 1 H NMR and IR spectroscopy [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d5sc06866e]
- 50. Microanalytical Characterization of an Innovative Modern ... [https://www.mdpi.com/1420-3049/28/2/564]
- 51. A stable and high-contrast color/fluorescence dual-mode ... [https://www.sciencedirect.com/science/article/abs/pii/S1385894725023332]
- 52. The Power of FTIR Spectroscopy for Faster, Safer ... [https://specac.com/ftir-applications/the-power-of-ftir-spectroscopy-for-faster-safer-pharmaceutical-formulations/]
- 53. FTIR Spectroscopy for Pharmaceutical Formulation [https://www.azom.com/article.aspx?ArticleID=24704]
- 54. Crash Course in Fourier Transform Infrared Spectroscopy [https://bitesizebio.com/38095/crash-course-in-fourier-transform-infrared-spectroscopy/]
- 55. Fourier transform infrared spectrometry (FTIR) for ... [https://www.sciencedirect.com/science/article/abs/pii/S0003267097816066]
- 56. New FTIR Applications in Pharmaceutical Analysis [https://www.thermofisher.com/blog/materials/new-ftir-applications-in-pharmaceutical-analysis/]
- 57. Introduction to hyphenated techniques and their ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3658024/]
- 58. GPC–IR Hyphenated Technology to Characterize ... [https://www.spectroscopyonline.com/view/gpc-ir-hyphenated-technology-characterize-copolymers-and-deformulate-complex-polymer-mixtures-polyme]
- 59. Gas Chromatography-Infrared Spectroscopy (GC-IR) [https://www.solutions.bocsci.com/resources/gas-chromatography-infrared-spectroscopy-gc-ir.html]
- 60. Using Multivariate Curve Resolution to Identify the Evolved ... [https://www.spectroscopyonline.com/view/using-multivariate-curve-resolution-identify-evolved-gases-created-during-tga-ir-experiment]
- 61. LC–NMR for Natural Product Analysis: A Journey from an ... [https://www.mdpi.com/2413-4155/3/1/6]
- 62. Protocol for determining protein dynamics using FT-IR ... [https://www.sciencedirect.com/science/article/pii/S2666166723005543]
- 63. FT-IR Spectroscopy for Microplastic Classification [https://www.spectroscopyonline.com/view/ft-ir-spectroscopy-for-microplastic-classification]
- 64. A Meta-Analysis Indeed and Practical Approach [https://www.mdpi.com/2073-4441/16/22/3170]
- 65. Virtual screening and molecular dynamics reveal the ... [https://pubmed.ncbi.nlm.nih.gov/40892061/]
- 66. In silico analysis of human CYP1A1 protein and ... [https://link.springer.com/article/10.1007/s44371-025-00244-6]
- 67. Multivariate Curve Resolution Methodology Applied to the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10302750/]
- 68. More Theory and Practice: The Thorny Problem of Mixtures ... [https://www.spectroscopyonline.com/view/more-theory-and-practice-thorny-problem-mixtures-and-more-straight-chain-alkanes]
- 69. Unexpected Resonances Could Boost NMR's Potency [https://physics.aps.org/articles/v18/145]
- 70. Enhancing the Hyperpolarized Xenon NMR Signal through ... [https://archive.ismrm.org/2025/4643.html]
- 71. 2025 Pharma Trends: Structure elucidation services by NMR [https://resolvemass.ca/structure-elucidation-services-by-nmr/]
- 72. Achieving high resolution and optimizing in spatial... sensitivity [https://pubs.rsc.org/en/content/articlelanding/2016/cp/c6cp01054g]
- 73. Spectrometers Comparison : SHIMADZU (Shimadzu Corporation) FTIR [https://www.shimadzu.com/an/products/molecular-spectroscopy/ftir/comparison/index.html]
- 74. Analytical quality by design-compliant development of ... [https://www.sciencedirect.com/science/article/abs/pii/S0026265X24026572]
- 75. The Future of FT-IR Imaging [https://www.spectroscopyonline.com/view/the-future-of-ft-ir-imaging]
- 76. NMR Chemical Shifts of Impurities [https://www.sigmaaldrich.com/US/en/technical-documents/technical-article/analytical-chemistry/nuclear-magnetic-resonance/1h-nmr-and-13c-nmr-chemical-shifts-of-impurities-chart?srsltid=AfmBOoqfmOwKggduK1yOp1ykSDsjx6fJNYkMwXly0rUE0aIylzbwURLN]
- 77. Machine learning-guided ATR-FTIR for in-depth analysis of ... [https://www.sciencedirect.com/science/article/abs/pii/S0925963525004091]
- 78. Qualitative analysis of edible oil mixture for omega-3 ... [https://www.nature.com/articles/s41538-025-00500-0]
- 79. Distinguishing Impurities … Part 1 [https://www.acdlabs.com/blog/when-impurities-go-bad-part-1/]
- 80. Application of two- and three-way chemometric methods in ... [https://www.sciencedirect.com/science/article/abs/pii/S0003267000007078]
- 81. "Spectral deconvolution of chromatograms without offline analytics ... [https://dc.engconfintl.org/biomanufact_ii/95/]
- 82. Application of Chemometrics Approaches to Analysis ... [https://pubmed.ncbi.nlm.nih.gov/30058366/]
- 83. Spectroscopic properties (FT-IR, NMR and UV) and DFT ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10709190/]
- 84. FTIR as a Method for Qualitative Assessment of Solid ... [https://www.mdpi.com/1420-3049/27/24/8846]
- 85. FT-IR Spectroscopy Mini-Tutorial: Principles, Practice, and ... [https://www.spectroscopyonline.com/view/ft-ir-spectroscopy-mini-tutorial-principles-practice-and-applications-across-disciplines]
- 86. Exploring the Frontiers of Computational NMR: Methods ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12530207/]
- 87. 3.2. Spectral Analysis using NIRS, FTIR, and NMR [https://bio-protocol.org/exchange/minidetail?id=9255483&type=30]
- 88. Full Length Article FTIR, 1 H-/ 13 C-NMR spectral ... [https://www.sciencedirect.com/science/article/pii/S2667022424002974]
- 89. How an FTIR Spectrometer Operates [https://chem.libretexts.org/Bookshelves/Physical_and_Theoretical_Chemistry_Textbook_Maps/Supplemental_Modules_(Physical_and_Theoretical_Chemistry)/Spectroscopy/Vibrational_Spectroscopy/Infrared_Spectroscopy/How_an_FTIR_Spectrometer_Operates]
- 90. 60 MHz 1H NMR spectroscopy for the analysis of edible oils [https://pmc.ncbi.nlm.nih.gov/articles/PMC4024201/]
- 91. Direct Stoichiometry, FTIR, and 1 H-NMR [https://www.mdpi.com/2304-8158/9/1/83]
- 92. Fourier transform infrared spectroscopic technique for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12426772/]
- 93. Structure Analysis [https://www.bruker.com/en/applications/pharma/drug-discovery/structure-analysis.html]
- 94. Overcoming the challenge of quantifying aged microplastic by qNMR... [https://pubs.rsc.org/en/content/articlelanding/2025/em/d5em00393h]
- 95. Top Quantitative (qNMR) Companies... Nuclear Magnetic Resonance [https://www.linkedin.com/pulse/top-quantitative-nuclear-magnetic-resonance-qnmr-companies-vzqnc]
- 96. FTIR accessories for pharmaceutical analysis [https://specac.com/ftir-applications/are-you-missing-contaminants-why-ftir-is-still-a-must-have-in-pharma-qc/]
- 97. 1H NMR, FAAS, portable NIR, benchtop NIR, and ATR- ... [https://www.sciencedirect.com/science/article/abs/pii/S0169743923001570]
- 98. Combining Mass Spectrometry and NMR Improves Metabolite ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6668615/]
- 99. Cross-validation (statistics) [https://en.wikipedia.org/wiki/Cross-validation_(statistics)]
- 100. A comprehensive review-current development in ... [https://www.sciencedirect.com/science/article/pii/S2211715625003248]
- 101. High-Resolution NMR for Complex Molecule Analysis [https://www.creative-biostructure.com/resource-high-resolution-nmr-complex-molecule-analysis.htm?srsltid=AfmBOor6Y4jXPzo39c7sQmqsnrc4dCkq_FGXHsQqSHGyEqnVZQnu0L-n]
- 102. NMR methods for the analysis of mixtures - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9753098/]