Measurement Uncertainty in Pharmaceutical Analysis: A Comprehensive Comparison of UV Spectroscopy and HPLC
This article provides a detailed comparison of measurement uncertainty between UV spectroscopy and High-Performance Liquid Chromatography (HPLC) for researchers, scientists, and drug development professionals.
Measurement Uncertainty in Pharmaceutical Analysis: A Comprehensive Comparison of UV Spectroscopy and HPLC
Abstract
This article provides a detailed comparison of measurement uncertainty between UV spectroscopy and High-Performance Liquid Chromatography (HPLC) for researchers, scientists, and drug development professionals. It explores the fundamental principles governing uncertainty in each technique, examines their methodological applications across pharmaceutical testing scenarios, and offers practical strategies for troubleshooting and optimization. By synthesizing foundational concepts with validation data and contemporary case studies, this review delivers critical insights for selecting the appropriate analytical method based on fitness-for-purpose, regulatory requirements, and uncertainty considerations in biomedical research and quality control environments.
Core Principles and Sources of Uncertainty in UV and HPLC
In the realm of pharmaceutical analysis, UV-Vis spectrophotometry and High-Performance Liquid Chromatography (HPLC) represent two fundamental analytical approaches with distinct operating principles and applications. UV-Vis spectrophotometry is a technique that measures the absorption of ultraviolet or visible light by a compound in solution, following the Beer-Lambert law which states that absorbance is proportional to the concentration of the absorbing species [1]. This method provides a straightforward approach for quantitative analysis but lacks inherent separation capabilities, making it susceptible to interference from other absorbing compounds in the sample.
In contrast, HPLC is a sophisticated separation technique that utilizes a liquid mobile phase to force compounds through a column packed with solid stationary phase particles. Separation occurs based on differential partitioning of analytes between the mobile and stationary phases, followed by detection, typically using UV-Vis spectrophotometry [2]. This two-stage process (separation followed by detection) gives HPLC its superior selectivity compared to direct spectrophotometric measurements. The fundamental distinction lies in HPLC's ability to physically separate mixture components before quantification, thereby reducing interference and improving analytical specificity for complex samples [2].
Measurement Uncertainty: A Comparative Framework
Measurement uncertainty is a critical parameter that quantifies the doubt associated with an analytical result, defining the range within which the true value is expected to lie [3]. Understanding and comparing the uncertainty profiles of UV-Vis spectrophotometry and HPLC is essential for selecting the appropriate method for pharmaceutical analysis.
UV-Vis spectrophotometry encounters multiple uncertainty sources that can be categorized into instrumental and procedural factors. Instrumental uncertainties include spectrophotometer reading repeatability, instrumental drift, wavelength accuracy, stray light, and cell (cuvette) variability [4]. These are often considered "physical" sources of uncertainty originating from the instrument itself.
Perhaps more significantly, "chemical" uncertainty sources often contribute more substantially to the combined uncertainty. These include interference from matrix constituents, incomplete extraction of the analyte from the sample, decomposition of the photometric complex, non-linearity deviations from the Beer-Lambert law, and operator effects [4]. The lack of separation capability means that any component in the sample matrix with overlapping absorption characteristics will contribute to the measured absorbance, potentially leading to significant positive bias in results.
In HPLC, the uncertainty structure is more complex due to the multi-step nature of the technique, encompassing injection, separation, detection, and signal processing stages [5]. The injection step contributes uncertainty through the random variability of injected sample volume and concentration. The separation process introduces uncertainties related to retention volume, number of theoretical plates, temperature fluctuations, and flow rate variations [5].
Detection by spectrophotometry adds the same uncertainties inherent to UV-Vis measurements, compounded by the fact that the concentration arriving at the detector has already been affected by separation uncertainties. Simulation studies have shown that uncertainty in injection volume typically contributes the greatest uncertainty to the final result in HPLC analysis [5]. However, the separation step significantly reduces uncertainties related to matrix interference compared to direct spectrophotometry.
Comparative Uncertainty Profiles
Table 1: Comparative Uncertainty Profiles of UV-Vis Spectrophotometry and HPLC
| Uncertainty Factor | UV-Vis Spectrophotometry | HPLC |
|---|---|---|
| Matrix Interference | High susceptibility | Minimal due to separation |
| Instrumental Noise | Moderate impact | Moderate impact |
| Injection Volume | Not applicable | Major contribution |
| Sample Preparation | High impact | Moderate impact |
| Flow Rate Variability | Not applicable | Significant impact |
| Specificity | Low (depends on clean spectra) | High (separation-based) |
| Linearity Range | Generally wide | Wide but detector-dependent |
The fundamental difference in uncertainty profiles stems from HPLC's ability to separate analytes from interfering substances, thereby eliminating what is often the largest uncertainty component in direct spectrophotometric measurements [4] [2]. This makes HPLC particularly advantageous for analyzing complex mixtures like pharmaceutical formulations, where excipients and degradation products may interfere with the target analyte.
Diagram 1: Uncertainty sources in UV-Vis spectrophotometry versus HPLC. Note the more complex uncertainty structure in HPLC, but reduced matrix interference.
Experimental Protocols and Methodologies
UV-Vis Spectrophotometric Method Protocol
The development and validation of a UV-Vis spectrophotometric method for pharmaceutical analysis follows a standardized protocol. For the analysis of repaglinide, as described in one study, the methodology involves dissolving the drug in methanol and measuring absorbance at 241 nm [1]. The specific experimental workflow includes:
Standard Solution Preparation: A primary stock solution of 1000 μg/mL is prepared in methanol. Working standard solutions are prepared by appropriate dilution to cover the concentration range of 5-30 μg/mL [1].
Sample Preparation: For tablet formulations, twenty tablets are weighed to determine mean weight and finely powdered. A portion equivalent to 10 mg of active ingredient is dissolved in 30 mL methanol in a 100 mL volumetric flask, sonicated for 15 minutes, diluted to volume, and filtered [1].
Analysis Procedure: The absorbance of prepared solutions is measured against a methanol blank at the predetermined wavelength (241 nm for repaglinide). The concentration is determined from a calibration curve constructed using standard solutions [1].
Method validation parameters include linearity (r² > 0.999 for repaglinide across 5-30 μg/mL), precision (RSD < 1.5%), and accuracy with mean recoveries of 99.63-100.45% [1].
HPLC Method Protocol
A typical reversed-phase HPLC method for pharmaceutical analysis follows a more complex protocol due to the separation requirements. For repaglinide analysis, the methodology includes:
Chromatographic Conditions:
- Column: Agilent TC-C18 (250 mm × 4.6 mm i.d., 5 μm particle size)
- Mobile Phase: Methanol and water (80:20 v/v, pH adjusted to 3.5 with orthophosphoric acid)
- Flow Rate: 1.0 mL/min
- Detection: UV at 241 nm
- Injection Volume: 20 μL [1]
Standard Solution Preparation: A primary stock solution of 1000 μg/mL is prepared in methanol. Working standard solutions are prepared by dilution with mobile phase to cover the concentration range of 5-50 μg/mL [1].
Sample Preparation: Tablet powder equivalent to 10 mg of active ingredient is dissolved and extracted with methanol, followed by dilution with mobile phase to obtain final concentrations within the linearity range [1].
System Suitability Testing: Before analysis, system suitability parameters are verified, including peak symmetry (tailing factor: 1.22 for repaglinide) and retention time reproducibility [1].
Method validation demonstrates linearity (r² > 0.999 across 5-50 μg/mL), precision (RSD < 1.5%), and accuracy with mean recoveries of 99.71-100.25% [1].
Diagram 2: Comparative workflows for UV-Vis spectrophotometry and HPLC analysis.
Performance Comparison and Experimental Data
Quantitative Performance Metrics
Table 2: Comparative Analytical Performance of UV-Vis Spectrophotometry and HPLC
| Performance Parameter | UV-Vis Spectrophotometry | HPLC |
|---|---|---|
| Linearity Range | 5-30 μg/mL (repaglinide) [1] | 5-50 μg/mL (repaglinide) [1] |
| Correlation Coefficient (r²) | >0.999 [1] | >0.999 [1] |
| Precision (RSD) | <1.5% [1] | <1.5% [1] |
| Accuracy (% Recovery) | 99.63-100.45% [1] | 99.71-100.25% [1] |
| Limit of Detection | Higher (matrix-dependent) | Lower (separation reduces noise) |
| Analysis Time | Minutes | 10-30 minutes per run |
| Specificity | Limited without separation | High with optimal separation |
| Multi-analyte Capability | Limited without chemometrics | Excellent |
Case Study: Bakuchiol in Cosmetic Products
A comparative study analyzing bakuchiol in cosmetic products demonstrated the practical implications of technique selection. UV-Vis analysis was performed at 262 nm in ethanol, but samples 5 and 6 (oil-in-water emulsions) could not be completely dissolved, preventing proper extraction and quantification of bakuchiol [6]. This highlights a significant limitation of direct spectrophotometry for complex matrices.
In contrast, HPLC analysis using a reversed-phase C18 column with isocratic elution (acetonitrile with 1% formic acid) and detection at 260 nm successfully quantified bakuchiol across all sample types [6]. The separation capability allowed for precise quantification without interference from emulsion components, with relative standard deviation for intraday variation below 2.5% [6].
Case Study: Simultaneous Analysis of Amlodipine and Benazepril
Another illustrative case involves the simultaneous determination of amlodipine besylate (AM) and benazepril hydrochloride (BZ) in fixed-dose combinations. A significant analytical challenge arose because both compounds absorb at 237 nm, making direct UV spectrophotometric quantification impossible without mathematical corrections [7].
Researchers developed a correlation equation approach using absorbance measurements at two wavelengths (237 nm and 366 nm) to resolve the overlapping spectra [7]. While this UV method provided a "cheap, reliable and less time consuming alternative," it required sophisticated mathematical treatment and still faced limitations in specificity compared to HPLC [7].
The developed HPLC method employed a Shodex C-18 column with a mobile phase of potassium dihydrogen phosphate buffer (pH 5.3) and acetonitrile (55:45 v/v), successfully separating AM (4.43 min) and BZ (5.70 min) without mathematical manipulation [7]. Both methods showed good correlation (r² > 0.999), but HPLC provided inherent separation and individual quantification without algorithmic intervention [7].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential Research Reagents and Materials for Analytical Methods
| Item | Function | Application in UV-Vis | Application in HPLC |
|---|---|---|---|
| HPLC-Grade Solvents | High purity mobile phase components | Sample dissolution [1] | Mobile phase constituent [1] |
| Buffer Salts | pH control of mobile phase | Limited use | Essential for retention time control [7] |
| C18 Chromatographic Columns | Stationary phase for separation | Not applicable | Essential for reversed-phase separation [1] [7] |
| Standard Reference Materials | Method calibration and validation | Quantitative calibration [1] | System calibration and peak identification [1] |
| Syringe Filters | Sample clarification | Required for turbid solutions [1] | Essential to protect HPLC column [1] |
| Volumetric Glassware | Precise solution preparation | Critical for standard and sample preparation [1] | Critical for mobile phase and sample preparation [1] |
The comparative analysis of UV-Vis spectrophotometry and HPLC reveals a clear trade-off between simplicity and specificity. UV-Vis spectrophotometry offers rapid, cost-effective analysis with minimal equipment requirements, making it suitable for simple matrices where target analytes have distinct absorption characteristics and minimal interference [1]. However, its susceptibility to matrix effects and limited specificity represent significant constraints for complex samples.
HPLC, with its separation capability before detection, provides superior specificity, lower detection limits, and robust performance across diverse sample matrices [2]. The primary disadvantages include higher instrumentation costs, longer analysis times, and more complex method development and validation requirements.
The choice between these techniques ultimately depends on the analytical problem: UV-Vis spectrophotometry serves well for routine quality control of simple formulations, while HPLC is indispensable for complex mixtures, stability-indicating methods, and research applications requiring high specificity. Understanding their fundamental operating principles and uncertainty profiles enables scientists to select the optimal approach for their specific analytical needs in pharmaceutical development and quality control.
In pharmaceutical analysis and drug development, the choice of analytical technique directly impacts the reliability, accuracy, and regulatory acceptance of data. Measurement uncertainty—a quantitative parameter characterizing the dispersion of values attributed to a measured quantity—serves as a critical indicator of analytical method performance [8]. Within a broader thesis comparing measurement uncertainty between UV spectroscopy and high-performance liquid chromatography (HPLC), this guide objectively examines the fundamental limitations of UV spectroscopy. Understanding these uncertainty sources is essential for researchers and scientists to make informed methodological choices, particularly when data quality directly influences drug safety and efficacy decisions.
UV spectroscopy operates on the principle of measuring the absorption of ultraviolet or visible light by analyte molecules, following the Beer-Lambert law which states that absorbance (A) is proportional to concentration (c), path length (l), and the molar absorptivity (ε) of the analyte [9]. Despite its simplicity, speed, and cost-effectiveness, UV spectroscopy exhibits specific vulnerability to chemical interferences and calibration limitations that significantly contribute to measurement uncertainty, especially when compared to more sophisticated separation-based techniques like HPLC [10] [11].
The measurement uncertainty in UV spectroscopy arises from both instrumental factors and sample-dependent characteristics. The key sources can be categorized into chemical interferences, calibration limitations, and instrumental parameters, each contributing differently to the overall uncertainty budget.
Chemical Interferences and Selectivity Limitations
Lack of Specificity: The fundamental limitation of UV spectroscopy lies in its low specificity, as any compound containing chromophores that absorb light at the selected wavelength will contribute to the measured absorbance signal [11]. This becomes particularly problematic in complex mixtures where excipients, impurities, or degradation products absorb at similar wavelengths as the target analyte, leading to positive interference and overestimation of concentration [10].
Matrix Effects: Real-world samples often contain multiple absorbing components, resulting in spectral overlap that UV spectroscopy cannot resolve without prior separation [12]. For instance, in the analysis of piperine in black pepper, UV spectroscopy demonstrated higher measurement uncertainty (4.29%) compared to HPLC (2.47%), primarily due to potential matrix interferences that HPLC could separate through chromatographic resolution [8].
Solvent and Environmental Factors: The UV absorbance of compounds can be influenced by solvent polarity, pH, temperature, and ionic strength, which may alter the absorption spectrum and introduce additional uncertainty if not carefully controlled [9].
Calibration and Linear Range Limitations
Non-Linearity Deviations: While the Beer-Lambert law assumes a linear relationship between absorbance and concentration, real-world measurements often deviate from linearity at higher absorbance values (typically above 1.0 AU) due to instrumental factors such as stray light or polychromatic radiation [9]. This necessitates careful validation of the calibration curve's linear range for each application.
Dynamic Range Constraints: UV spectroscopy offers a relatively limited dynamic range compared to HPLC, often requiring sample dilution or concentration to bring measurements within the optimal absorbance range (0.2-0.8 AU), introducing additional preparation steps and potential dilution errors [9].
Photometric Accuracy: The accuracy of absorbance measurements is influenced by instrumental factors including spectral bandwidth, stray light, wavelength accuracy, and detector linearity [13]. Stray light—radiation outside the nominal bandwidth—becomes a particularly significant error source at high absorbance values, leading to non-linear response and underestimated concentrations [13].
The following table summarizes the key uncertainty contributors and their impacts on analytical results:
Table 1: Critical Uncertainty Sources in UV Spectroscopy and Their Impacts
| Uncertainty Category | Specific Source | Impact on Measurement | Typical Magnitude |
|---|---|---|---|
| Chemical Interferences | Spectral overlap from multiple chromophores | False elevation of absorbance reading | Varies with matrix complexity |
| Solvent background absorption | Baseline offset and reduced sensitivity | Significant for UV-absorbing solvents | |
| pH-dependent spectral shifts | Wavelength maxima shifting | ±2-5 nm with pH variation | |
| Calibration Limitations | Stray light at high absorbance | Non-linearity in Beer-Lambert relationship | >0.5% stray light ratio [13] |
| Photometric non-linearity | Deviation from ideal calibration slope | ±1-2% in high-quality instruments [13] | |
| Limited dynamic range | Need for sample dilution/concentration | Additional 1-3% uncertainty [9] | |
| Instrumental Parameters | Wavelength inaccuracy | Shift from absorption maximum | ±0.5-2 nm depending on calibration [13] |
| Bandwidth selection | Reduced resolution and potential spectral overlap | 1-5 nm typically | |
| Cell pathlength variation | Direct effect on calculated concentration | ±0.01 mm for standard 10 mm cells |
Comparative Experimental Data: UV Spectroscopy vs. HPLC
Direct comparison studies provide compelling evidence for the performance differences between UV spectroscopy and HPLC, particularly in measurement uncertainty and reliability.
Piperine Quantification in Black Pepper
A rigorous 2022 methodology comparison study quantified the performance differences between UV spectroscopy and HPLC-UV for determining piperine content in black pepper [8]. The experimental protocol involved:
- Sample Preparation: Black pepper samples were ground, sieved through a 60-mesh screen, and extracted using appropriate solvents followed by filtration [8].
- UV Methodology: Absorbance measurements were performed at the wavelength of maximum absorption for piperine after establishing linearity in the range of 0.8-200 mg/kg [8].
- HPLC Methodology: Separation employed a C18 column with UV detection, using a mobile phase system optimized for piperine resolution from other matrix components [8].
- Validation Parameters: Both methods were validated for specificity, linearity, accuracy, precision, LOD, LOQ, and measurement uncertainty according to ICH guidelines [8].
Table 2: Performance Comparison of UV Spectroscopy and HPLC for Piperine Analysis [8]
| Validation Parameter | UV Spectroscopy | HPLC-UV |
|---|---|---|
| Linearity (R²) | Good | Good |
| Limit of Detection | 0.65 mg/kg | 0.23 mg/kg |
| Accuracy Range | 96.7 - 101.5% | 98.2 - 100.6% |
| Precision (RSD) | 0.59 - 2.12% | 0.83 - 1.58% |
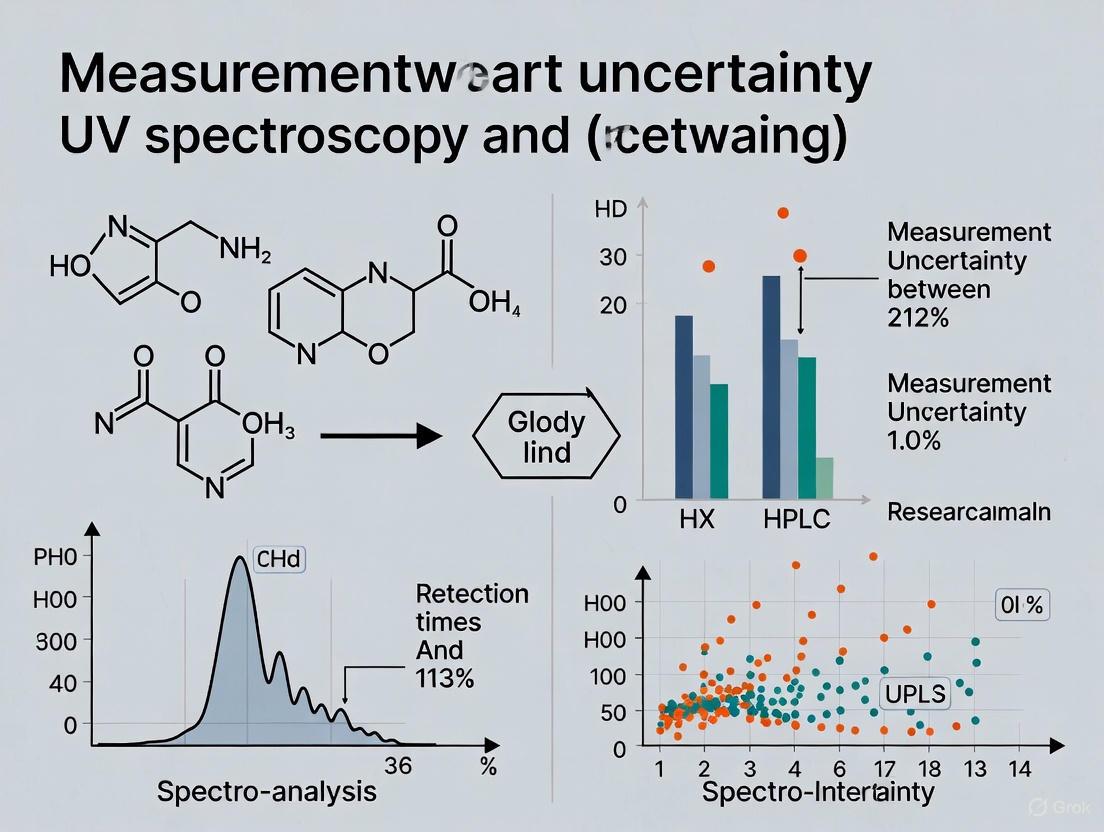
| Measurement Uncertainty | 4.29% (at 49.481 g/kg) | 2.47% (at 34.819 g/kg) |
| Key Uncertainty Sources | Matrix interferences, specificity limitations | Calibration uncertainty, preparation variations |
The experimental outcomes demonstrated that HPLC provided superior sensitivity (approximately three times lower LOD) and significantly lower measurement uncertainty (2.47% vs. 4.29%) compared to UV spectroscopy [8]. The primary advantage of HPLC emerged in its specificity—the chromatographic separation effectively isolated piperine from other pepper constituents that would otherwise contribute to collective UV absorbance in the direct spectroscopic method [8].
Nanoplastics Quantification
A 2025 environmental study evaluated UV-visible spectroscopy for quantifying true-to-life nanoplastics and benchmarked it against mass-based techniques including pyrolysis GC-MS and thermogravimetric analysis [12]. The experimental workflow included:
- Sample Preparation: Polystyrene-based nanoplastics were generated from fragmented plastic items through mechanical fragmentation under cryogenic conditions followed by sequential centrifugations to isolate the nanoplastic fraction [12].
- UV-Vis Methodology: Microvolume UV-vis spectroscopy was employed to minimize sample requirement and enable sample recovery for subsequent analyses [12].
- Comparative Techniques: Mass-based quantification using Py-GC-MS and TGA provided reference values for method comparison [12].
Despite some underestimation of nanoplastic concentrations relative to mass-based techniques, UV-vis spectroscopy provided consistent results in terms of order of magnitude and showed reliable trends across different methods [12]. The study concluded that UV-vis spectroscopy offers a rapid, accessible, and effective means of quantifying nanoplastics, particularly when sample volumes are limited, though with higher uncertainty compared to reference mass-based methods [12].
Method Workflows and Visualization
The analytical workflow differs significantly between UV spectroscopy and HPLC, largely accounting for their differing vulnerability to chemical interferences and measurement uncertainty. The following diagram illustrates the fundamental processes of each technique:
Diagram 1: Analytical technique workflows compared
The critical distinction lies in HPLC's separation step, which physically isolates the analyte from potential interferents before detection, thereby eliminating one of the most significant uncertainty sources that plague direct UV spectroscopy measurements [10] [11].
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful implementation of either analytical technique requires specific materials and reagents, each contributing differently to measurement uncertainty:
Table 3: Essential Research Materials and Their Functions
| Material/Reagent | Function in Analysis | Uncertainty Considerations |
|---|---|---|
| High-Purity Solvents | Dissolve samples and standards; HPLC mobile phase | UV-cutoff wavelength affects baseline noise; purity impacts detector response |
| Certified Reference Standards | Calibration curve establishment; method validation | Purity uncertainty directly transfers to systematic measurement bias |
| Cuvettes (UV-Vis) | Contain sample for light absorption measurement | Pathlength tolerance (±0.01 mm) contributes directly to concentration uncertainty |
| HPLC Columns | Stationary phase for compound separation | Column efficiency and selectivity determine resolution from interferents |
| Buffer Components | pH control and ionic strength modification | pH-sensitive absorbance changes introduce spectral shift uncertainty |
| Filters (0.45 μm or 0.22 μm) | Particulate removal from samples | Filter adsorption losses can reduce apparent concentration |
Implications for Analytical Method Selection
The choice between UV spectroscopy and HPLC involves balancing multiple factors including analytical requirements, resource constraints, and regulatory considerations.
Appropriate Applications for UV Spectroscopy
- Simple Mixtures: UV spectroscopy remains reliable for analyzing single-component systems or simple mixtures where potential interferents are known to be absent or non-absorbing at the measurement wavelength [11].
- Routine Quality Control: In pharmaceutical quality control, UV spectroscopy finds appropriate application in raw material identification, dissolution testing of immediate-release formulations, and routine assay of drug substances when method specificity has been conclusively demonstrated [10].
- Limited Resource Settings: The technique's simplicity, speed, and lower operational cost make it valuable for screening analyses and settings with limited access to sophisticated instrumentation [10].
When HPLC Becomes Essential
- Complex Matrices: HPLC is unequivocally superior for analyzing complex biological samples, natural products, and multi-component formulations where chemical interferences would otherwise compromise UV spectroscopy results [8] [11].
- Regulatory Submissions: For pharmaceutical registration dossiers submitted to regulatory agencies (FDA, EMA), HPLC is typically required for assay and impurity profiling due to its superior specificity and lower measurement uncertainty [14] [11].
- Low Concentration Analytics: When analyzing trace-level compounds or impurities, HPLC provides significantly lower detection limits and superior sensitivity, particularly when coupled with mass spectrometric detection [10].
UV spectroscopy remains a valuable analytical technique with distinct advantages in simplicity, speed, and cost-effectiveness for appropriate applications. However, its inherent vulnerability to chemical interferences and calibration limitations introduces significant measurement uncertainty compared to HPLC, particularly for complex samples. The comparative experimental data demonstrates that HPLC typically provides 40-50% lower measurement uncertainty than UV spectroscopy for complex matrix analysis [8].
For researchers and drug development professionals, the selection between these techniques should be guided by the sample complexity, analytical requirements, and necessary data quality. When the highest level of accuracy and specificity is required—particularly for regulatory submissions or complex biological samples—HPLC's separation power provides unequivocal advantages in uncertainty reduction. UV spectroscopy serves as an efficient screening tool or routine method for well-characterized simple systems, but its limitations must be respected through adequate method validation and uncertainty estimation to ensure data reliability.
In the rigorous world of pharmaceutical analysis, measurement uncertainty is an omnipresent factor that quantifies the doubt associated with any analytical result. For researchers and drug development professionals, understanding and controlling these uncertainties is not merely a technical exercise but a fundamental requirement for ensuring product quality, safety, and efficacy, as well as for meeting regulatory standards. Both High-Performance Liquid Chromatography (HPLC) and UV spectroscopy are cornerstone techniques in pharmaceutical analysis, yet they exhibit distinct uncertainty profiles and are susceptible to different error sources. HPLC, with its separation capabilities, generally offers superior specificity for complex mixtures but involves more procedural steps where errors can accumulate. UV spectroscopy, while simpler and faster, often lacks selectivity when analyzing complex samples, potentially leading to interference-related inaccuracies.
This guide objectively compares the performance of these two techniques by examining the primary sources of uncertainty, supported by experimental data and structured within a framework that aligns with regulatory guidelines such as ICH Q2(R2) and quality standards like those in the United States Pharmacopeia (USP). A critical understanding of these factors enables scientists to select the most appropriate method, implement effective control strategies, and accurately interpret their analytical results, thereby strengthening the entire drug development pipeline from formulation screening to quality control.
Core Uncertainty Contributors: HPLC vs. UV Spectroscopy
The reliability of an analytical result is fundamentally determined by how uncertainties from various stages of the method combine. The table below provides a comparative overview of the primary uncertainty sources in HPLC and UV spectroscopy.
Table 1: Core Uncertainty Contributors in HPLC and UV Spectroscopy
| Analysis Phase | Uncertainty Contributor | Impact on HPLC | Impact on UV Spectroscopy |
|---|---|---|---|
| Sample Preparation | Weighing, Dilution, Extraction | Major contributor, especially for complex matrices [15] [14] | Significant, often the dominant source due to simpler overall workflow [10] |
| Calibration | Reference Standard Purity, Curve Fitting | Significant; model choice (e.g., ordinary vs. weighted least squares) affects uncertainty [5] | Significant; relies on accurate standards and linearity across range [10] |
| Instrumental Analysis | Injection Volume | A major instrumental contributor; random uncertainty in autosampler delivery [5] | Not applicable (uses cuvettes of fixed pathlength) |
| Flow Rate | Contributes to retention time and peak shape uncertainty [5] | Not applicable | |
| Signal Detection & Processing | Detector Noise (UV/Vis) | Contributes to peak area/height uncertainty [5] | A primary source of noise, directly impacting absorbance measurement [10] |
| Peak Integration | A well-documented source of error, especially for small or poorly resolved peaks [16] | Not applicable | |
| Selectivity | Matrix Interference | High separation capability generally reduces impact [10] | Often a major limitation; overlapping spectra can cause significant bias [10] |
Uncertainty from Sampling and Sample Preparation
In both techniques, sample preparation is a critical source of uncertainty. For HPLC, the multi-step process—involving weighing, dilution, and often extraction—introduces cumulative errors in mass and volume. A bottom-up approach to uncertainty assessment, as outlined in guides from ISO GUM and EURACHEM, quantifies these contributions from homogeneity, precision, and sample preparation [15] [17]. In UV spectroscopy, while the preparation may be simpler, the same principles of weighing and dilution uncertainty apply. Because the overall method is less complex, these preparation errors can constitute a larger relative contribution to the final result's uncertainty budget [10].
Uncertainty from Calibration
The calibration process is a significant fountainhead of uncertainty in quantitative analysis. In HPLC, the relationship between peak area/height and concentration is not the only consideration. Research indicates that the concentration of the analyte at the detector is not zero, even if the injected standard concentration is perfectly known; it carries uncertainty propagated from the injection volume and chromatographic separation process [5]. This makes calibration models that account for errors in both variables (x and y) sometimes more appropriate than classical least squares.
For UV spectroscopy, the calibration curve is also central, built from the absorbance readings of known standards. Uncertainty arises from the purity of the reference standard, the accuracy of dilutions, and the instrument's baseline noise affecting absorbance measurements [10]. The linearity of this curve across the working range must be rigorously validated, as any non-linearity introduces bias and additional uncertainty.
Uncertainty from Peak Integration in HPLC
Peak integration is a uniquely challenging source of uncertainty in chromatographic methods. Automated data systems can miscalculate peak areas due to various baseline anomalies. Common errors include:
- Negative Peaks or Baseline Dips: The system may mistakenly identify a dip as the baseline start, leading to incorrect area assignment [16].
- Poorly Resolved Peaks: The choice between a perpendicular drop and a skimming method (tangent) to split two overlapping peaks is critical. Misapplication can drastically misrepresent impurity levels; for instance, a perpendicular drop can assign more than twice the correct area to a minor peak, potentially causing a batch to fail quality specifications [16].
- Noisy or Drifting Baselines: Small peaks are particularly vulnerable to incorrect baseline placement by the integration algorithm, either starting too early or ending too late [16].
These scenarios often necessitate manual reintegration, which is permitted under strict guidelines (e.g., CFR 21 Part 11) that require the original data to be preserved, the user to be identified, and a valid reason for the change to be documented [16].
Experimental Comparison and Uncertainty Data
Direct, side-by-side method comparisons provide the most concrete evidence for evaluating uncertainty. The following table summarizes key validation parameters from a study on the antidiabetic drug repaglinide, offering a quantitative performance comparison.
Table 2: Experimental Validation Data for Repaglinide Assay (UV vs. HPLC) [1]
| Validation Parameter | UV Spectroscopy | HPLC Method |
|---|---|---|
| Linearity Range | 5–30 μg/mL | 5–50 μg/mL |
| Correlation Coefficient (r²) | > 0.999 | > 0.999 |
| Precision (% R.S.D.) | < 1.50% | Highly precise (implied < R.S.D. of UV) |
| Accuracy (% Recovery) | 99.63–100.45% | 99.71–100.25% |
| Limit of Quantification (LOQ) | Not Specified | Not Specified |
| Key Advantages | Fast, economical, simple | Superior specificity, separation, precision |
The data demonstrates that both methods can be optimized to exhibit excellent linearity and accuracy. The critical difference lies in their operational range and precision. The HPLC method offers a wider linear range (5-50 μg/mL), making it more versatile for analyzing samples with a broad concentration spread. Furthermore, while the UV method is precise enough for many quality control applications, the HPLC method is noted for its high precision, which is crucial for detecting low-level impurities and ensuring batch-to-batch consistency [1]. This higher precision directly contributes to a tighter uncertainty budget for the HPLC result.
Detailed HPLC Experimental Protocol
The robust HPLC method for repaglinide can be summarized as follows [1]:
- Instrumentation: Agilent 1120 Compact LC with UV detector.
- Column: Agilent TC-C18 (250 mm × 4.6 mm, 5 μm).
- Mobile Phase: Methanol and water (80:20 v/v, pH adjusted to 3.5 with orthophosphoric acid).
- Flow Rate: 1.0 mL/min.
- Detection: 241 nm.
- Injection Volume: 20 μL.
- Sample Preparation: Tablets were powdered, dissolved in methanol, sonicated, filtered, and diluted with mobile phase.
This method resulted in a peak with good symmetry (tailing factor of 1.22) and a short run time, confirming its suitability for routine analysis [1]. The careful adjustment of the mobile phase pH is a critical robustness measure that helps control retention time variability, a potential uncertainty contributor.
Detailed UV Spectroscopy Experimental Protocol
The comparative UV method was executed as follows [1]:
- Instrumentation: Shimadzu 1700 Double beam UV-Vis spectrophotometer with 1.0-cm quartz cells.
- Wavelength: 241 nm.
- Solvent: Methanol.
- Sample Preparation: Tablets were powdered, dissolved in methanol, sonicated, filtered, and diluted with methanol.
- Procedure: The absorbance of the prepared sample and standards was measured against a methanol blank.
The simplicity of this protocol is its greatest strength, but it also introduces a key limitation: any component in the tablet formulation that also absorbs at 241 nm will contribute to the signal, causing a positive bias and increasing measurement uncertainty [10].
Visualization of Uncertainty Relationships and Workflows
Understanding how different uncertainty sources interact is key to managing them. The following diagram maps the logical relationships and propagation of uncertainty in an HPLC-UV analysis.
The workflow for quantifying uncertainty, especially using a bottom-up approach, can be systematically implemented. The following chart outlines the key steps in this process, which aligns with international standards.
The Scientist's Toolkit: Essential Research Reagents and Materials
The following table details key reagents, materials, and software solutions essential for conducting reliable HPLC and UV analyses, along with their specific functions in method development and uncertainty control.
Table 3: Essential Research Reagents and Solutions for HPLC and UV Analysis
| Category | Item | Primary Function | Uncertainty Consideration |
|---|---|---|---|
| Chromatography | C18 Column (e.g., Agilent TC-C18) | Stationary phase for analyte separation | Column batch-to-batch variability affects retention time. |
| HPLC-Grade Solvents (Methanol, Water) | Mobile phase components | Purity is critical to avoid baseline noise and ghost peaks. | |
| Reference Standards (e.g., Repaglinide) | Calibration and quantification | Purity and stability are direct uncertainty contributors. | |
| Sample Prep | Volumetric Flasks, Pipettes | Precise dilution and preparation | Accuracy and calibration of this glassware define preparation uncertainty. |
| Syringe Filters | Clarification of sample solutions | Can adsorb analyte, leading to recovery uncertainty. | |
| Software & Tools | Open-Access MUCalc [15] | Bottom-up measurement uncertainty calculation | Implements GUM and EURACHEM guides for standardized estimation. |
| Chromatography Data System (CDS) | Peak integration and quantification | Algorithm settings and manual reintegration rules are key. |
In summary, the choice between HPLC and UV spectroscopy for pharmaceutical analysis involves a careful trade-off between complexity and specificity, directly impacting the measurement uncertainty profile. UV spectroscopy offers a straightforward, cost-effective path for simple assays but is highly vulnerable to uncertainty from spectral interferences. HPLC, while more complex and involving more potential error sources (notably in injection volume, calibration model, and peak integration), provides the separation power necessary for specific and accurate analysis of complex mixtures, such as in impurity profiling and stability-indicating methods.
For researchers, the imperative is to align the analytical technique with the product's critical quality attributes. The experimental data confirms that both methods can be validated to be accurate and precise. However, HPLC is unequivocally more suited for applications demanding high specificity. By systematically identifying, quantifying, and controlling the major contributors discussed—especially those from sampling, calibration, and for HPLC, peak integration—scientists can ensure their methods are not only compliant with regulatory standards but also fundamentally robust and reliable, thereby de-risking the drug development process.
In the realm of analytical chemistry, the choice between Ultraviolet-Visible (UV-Vis) spectroscopy and High-Performance Liquid Chromatography (HPLC) represents a fundamental trade-off between simplicity and specificity. For researchers, scientists, and drug development professionals, this decision directly impacts the reliability of analytical data, particularly when dealing with increasingly complex pharmaceutical formulations and biological matrices. The core distinction lies in each technique's inherent ability to discriminate target analytes from interfering substances—a characteristic formally recognized as analytical specificity.
UV spectroscopy operates on the principle of measuring absorbance of light in the ultraviolet and visible regions, generating spectra based on electronic transitions in chromophores. While excellent for pure compounds or simple mixtures, this technique struggles with complex samples where spectral overlapping occurs [18]. In contrast, HPLC incorporates a separation step prior to detection, physically resolving components in time and space before quantification. This additional dimension of separation grants HPLC superior specificity for complex mixtures, albeit with increased operational complexity and cost [5] [19].
The distinction becomes critically important in pharmaceutical analysis where regulatory requirements demand rigorous method validation including specificity assessments. As we examine experimental evidence across various applications, the performance gap between these techniques in handling simple versus complex matrices becomes quantitatively evident, providing a framework for informed analytical method selection.
Theoretical Foundations: Mechanisms of Detection and Separation
UV-Vis Spectroscopy Fundamentals
UV-Vis spectroscopy measures the attenuation of light passing through a sample solution, following the Beer-Lambert law which establishes a linear relationship between absorbance and analyte concentration. The technique captures electronic transitions occurring when molecules absorb energy in the 190-800 nm range, producing characteristic spectra based on molecular structure [1].
Key advantages of UV-Vis include operational simplicity, rapid analysis, minimal sample preparation requirements, and cost-effectiveness for routine analysis of single components. Modern UV spectrophotometers incorporate advanced features including diode-array configurations for full-spectrum capture, microvolume sampling, and chemometric software for basic multicomponent analysis [20]. However, the fundamental limitation remains: without physical separation, all UV-absorbing compounds in the light path contribute to the measured signal, creating potential for positive interference.
HPLC Separation Principles
HPLC separates complex mixtures using differential partitioning between a stationary phase (column packing material) and a mobile phase (liquid solvent system). Components migrate at different rates through the column based on their chemical properties, emerging as distinct peaks with characteristic retention times [19] [21].
The chromatographic process introduces multiple dimensions of selectivity: chemical nature of the stationary phase (C18, CN, phenyl, etc.), mobile phase composition (pH, organic modifier, buffers), temperature, and flow rate. When coupled with UV detection (HPLC-UV), this separation provides two orthogonal identification parameters—retention time and spectral characteristics—significantly enhancing specificity over stand-alone UV spectroscopy [5].
The fundamental distinction in analytical approach between these techniques creates a natural application boundary: UV for simple, well-characterized systems and HPLC for complex, multi-component matrices where resolution of individual components is essential for accurate quantification.
Experimental Evidence: Quantitative Performance Comparison
Direct Method Comparison Studies
Rigorous comparative studies provide quantitative evidence of performance differences between UV spectroscopy and HPLC across various applications. The data reveal consistent patterns in measurement uncertainty, accuracy, and sensitivity.
Table 1: Comparative Method Performance for Pharmaceutical Analysis
| Analyte | Matrix | Technique | Linearity (R²) | Recovery (%) | Precision (RSD%) | LOQ | Measurement Uncertainty |
|---|---|---|---|---|---|---|---|
| Levofloxacin [18] | SBF | UV-Vis | 0.9999 | 96.00-99.50 | NR | NR | NR |
| Levofloxacin [18] | SBF | HPLC | 0.9991 | 96.37-110.96 | NR | NR | NR |
| Repaglinide [1] | Tablet | UV-Vis | >0.999 | 99.63-100.45 | <1.50 | NR | NR |
| Repaglinide [1] | Tablet | HPLC | >0.999 | 99.71-100.25 | <1.50 | NR | NR |
| Piperine [8] | Black pepper | UV-Vis | NR | 96.7-101.5 | 0.59-2.12 | NR | 4.29% (k=2) |
| Piperine [8] | Black pepper | HPLC-UV | NR | 98.2-100.6 | 0.83-1.58 | NR | 2.47% (k=2) |
NR = Not Reported
For levofloxacin analysis in simulated body fluid, UV-Vis demonstrated superior linearity (R²=0.9999 vs. 0.9991 for HPLC), but showed more consistent recovery rates (96.00-99.50%) compared to the wider range observed with HPLC (96.37-110.96%) [18]. This pattern suggests that while UV can provide excellent performance for specific applications, HPLC may exhibit greater variability in certain matrices despite its superior separation capabilities.
Measurement Uncertainty and Sensitivity
A comprehensive study on piperine quantification in black pepper revealed significantly lower measurement uncertainty for HPLC-UV (2.47%) compared to UV spectroscopy (4.29%), demonstrating HPLC's superior reliability for complex natural product analysis [8]. The sensitivity advantage of HPLC is further evidenced by lower detection limits across multiple applications.
Table 2: Sensitivity Comparison for Different Applications
| Analyte | Matrix | Technique | LOD | LOQ | Linear Range |
|---|---|---|---|---|---|
| Piperine [8] | Black pepper | UV-Vis | 0.65 | NR | NR |
| Piperine [8] | HPLC-UV | 0.23 | NR | NR | |
| Quercetin [22] | Various foods | HPLC-UV | 0.15-0.31 mg/kg | 0.44-0.93 mg/kg | 0.5-50 mg/L |
| Antihypertensive drugs [19] | Pharmaceutical/Plasma | HPLC-UV | NR | NR | 0.1-25.6 μg/mL |
The consistently lower detection limits achievable with HPLC-UV systems provide superior capability for trace analysis, particularly valuable in bioavailability studies, impurity profiling, and analysis of active components in complex natural products [19] [8] [22].
Methodologies: Detailed Experimental Protocols
HPLC Method for Antihypertensive Drug Analysis
A validated HPLC-UV method for simultaneous determination of amlodipine, olmesartan, valsartan, and hydrochlorothiazide exemplifies approach for complex mixture analysis [19]:
Chromatographic Conditions:
- Column: ACE CN column (200 × 4.6 mm, 5 μm)
- Mobile Phase: Acetonitrile-methanol-10 mM phosphoric acid, pH 2.5 (7:13:80, v/v/v)
- Flow Rate: 1.0 mL/min
- Detection: UV at 235 nm
- Temperature: 30°C
- Injection Volume: 20 μL
Sample Preparation:
- Tablets powdered and extracted with methanol via mechanical shaking (20 min) and sonication (20 min)
- Extracts diluted with mobile phase and filtered before injection
- Plasma samples processed with liquid-liquid extraction prior to analysis
Validation Parameters:
- Linearity demonstrated over 0.1-25.6 μg/mL depending on analyte
- Precision RSD ≤6.1% for all compounds
- Accuracy: 89.4-105.8% for tablet analysis; 85.6-109.4% for plasma
This methodology highlights the comprehensive approach needed for reliable HPLC analysis of complex mixtures, with particular attention to sample preparation, chromatographic separation optimization, and rigorous validation.
UV Spectrophotometric Method for Repaglinide
A simplified UV method for repaglinide quantification in tablets illustrates the straightforward approach possible with UV spectroscopy for uncomplicated matrices [1]:
Instrumentation: Shimadzu 1700 Double beam UV-Vis spectrophotometer with 1.0 cm quartz cells
Analytical Conditions:
- Wavelength: 241 nm
- Solvent: Methanol
- Sample Preparation: Tablets powdered and dissolved in methanol with sonication
- Linearity Range: 5-30 μg/mL
Validation Results:
- Linearity: R² > 0.999
- Precision: RSD < 1.50%
- Accuracy: 99.63-100.45% recovery
The stark contrast in methodological complexity between these protocols underscores the different resource commitments and expertise required for each technique.
Analytical Workflow Comparison
The decision pathway for method selection between UV spectroscopy and HPLC involves multiple considerations including sample complexity, regulatory requirements, and available resources.
Essential Research Reagent Solutions
Successful implementation of either analytical approach requires specific reagents and materials optimized for each technique's requirements.
Table 3: Essential Research Reagents and Materials
| Item | Function | UV-Vis Applications | HPLC Applications |
|---|---|---|---|
| HPLC-Grade Solvents [19] [8] | Mobile phase preparation | Limited use | Critical for baseline stability and reproducibility |
| Buffer Salts (KH₂PO₄, etc.) [18] [19] | Mobile phase pH control | Optional for sample preparation | Essential for peak shape and retention |
| Analytical Standards [18] [1] [8] | Calibration and quantification | Required for quantitative work | Required for quantitative work |
| Solid Phase Extraction Cartridges [19] | Sample clean-up | Occasionally used | Frequently essential for complex matrices |
| Syringe Filters (0.45/0.22 μm) [8] | Particulate removal | Standard practice | Critical for column protection |
| HPLC Columns (C18, CN, etc.) [18] [19] | Compound separation | Not applicable | Essential for selectivity and resolution |
The reagent requirements for HPLC are substantially more specialized and extensive, contributing to higher operational costs but enabling analysis of complex mixtures not feasible with UV spectroscopy alone.
Applications in Pharmaceutical Analysis
Drug Formulation Analysis
For quality control of finished drug products, the choice between UV and HPLC depends on formulation complexity. Simple immediate-release tablets with single active ingredients may be suitably analyzed by UV spectroscopy, as demonstrated with repaglinide tablets showing excellent accuracy and precision [1]. However, fixed-dose combination products with multiple active ingredients typically require HPLC for independent quantification without interference, exemplified by antihypertensive combinations containing amlodipine, olmesartan/valsartan, and hydrochlorothiazide [19].
Biopharmaceutical Applications
The expanding biologics market has driven demand for high-sensitivity protein analytics, with modern UV instruments incorporating variable-pathlength cells for direct antibody concentration measurement up to 300 mg/mL without dilution [20]. However, characterization of complex biologics typically requires HPLC-based separation techniques coupled with advanced detection to assess purity, stability, and post-translational modifications.
Emerging Trends and Future Outlook
The analytical instrumentation landscape continues evolving, with notable trends impacting both UV and HPLC technologies. The UV spectroscopy market is experiencing growth in portable/hand-held devices (projected 7.46% CAGR), diode-array configurations (7.76% CAGR), and bioprocess monitoring applications (8.56% CAGR) [20]. Meanwhile, the HPLC market shows strong movement toward UHPLC systems offering higher resolution and faster analysis times, with the global analytical HPLC market projected to reach USD 7,800 million by 2033 [21].
Machine learning approaches are enhancing both techniques, with random forest regression models improving spectral prediction in GC-VUV applications [23], while advanced data analytics optimize HPLC method development and peak integration. These technological advancements continue to narrow the performance gap between techniques while expanding their respective application boundaries.
The specificity advantage of HPLC-UV for resolving complex mixtures is unequivocally demonstrated through quantitative performance metrics including lower measurement uncertainty, superior sensitivity, and reliable quantification in multi-component matrices. However, UV spectroscopy maintains significant utility for simple analytical challenges where its operational simplicity, rapid analysis time, and cost-effectiveness provide compelling advantages.
Informed technique selection requires systematic evaluation of sample complexity, regulatory requirements, and resource constraints. For drug development professionals, the trend toward increasingly complex formulations and combination therapies continues to expand the application domain for HPLC-based methods, while UV spectroscopy finds renewed utility in process analytical technology and bioprocess monitoring where rapid, in-line measurements provide value beyond ultimate specificity.
In metrology, measurement uncertainty is defined as the "expression of the statistical dispersion of the values attributed to a measured quantity." No measurement is exact, and when a quantity is measured, the outcome depends on the measuring system, the measurement procedure, the skill of the operator, the environment, and other effects. Even when measured repeatedly under the same conditions with a sufficiently resolved instrument, different values will generally be obtained each time. The dispersion of these values relates to how well the measurement is performed [24].
The international guide that defines measurement uncertainty is the "Guide to the Expression of Uncertainty in Measurement" (GUM), which has been adopted by national measurement institutes and international laboratory accreditation standards like ISO/IEC 17025. The GUM classifies the evaluation of uncertainty into two main types: Type A (evaluated by statistical methods) and Type B (evaluated by other means) [25] [24]. This classification serves to indicate two different ways of evaluating uncertainty components and is for convenience of discussion only—it does not indicate any difference in the nature of the components resulting from the two types of evaluation [25].
Understanding Type A and Type B Uncertainties
Type A Uncertainty (Random Error)
Type A Uncertainty is defined as the "evaluation of a component of measurement uncertainty by a statistical analysis of measured quantity values obtained under defined measurement conditions" [25]. In practice, this involves performing repeated measurements and analyzing the resulting data series statistically.
The standard evaluation of Type A uncertainty involves calculating:
- Arithmetic Mean: The central value of a set of numbers (( \bar{x} = \frac{\sum x_i}{n }))
- Standard Deviation: A measure of data dispersion from its mean (( s = \sqrt{\frac{\sum(x_i - \bar{x})^2}{n-1}} ))
- Degrees of Freedom: The number of values free to vary in the final calculation (typically n-1 for n measurements) [25]
Type A uncertainty is characterized by an observed frequency distribution. Following the Central Limit Theorem, as more samples are collected, the data increasingly resembles a normal distribution [25]. In experimental contexts, random errors manifest as unpredictable fluctuations in measurements caused by unknown changes in the experiment, instruments, or environmental conditions [26].
Type B Uncertainty (Systematic Error)
Type B Uncertainty is defined as the "evaluation of a component of measurement uncertainty determined by means other than a Type A evaluation of measurement uncertainty" [25]. This evaluation relies on scientific judgment using all relevant available information, which may include previous measurement data, experience with instruments, manufacturer's specifications, calibration certificates, and handbook reference data [27].
Unlike Type A, Type B uncertainty is characterized using an assumed probability distribution based on available information. Common probability distributions used in Type B evaluation include:
- Rectangular (Uniform) Distribution: Used when only the upper and lower limits of a value are known
- Triangular Distribution: Appropriate when values near the center of limits are more likely
- Normal Distribution: Applied when the uncertainty is stated with a given confidence level or based on known probabilities [27]
Systematic errors in experimental observations typically stem from measuring instruments or their wrong use by experimenters. These errors consistently bias measurements in a specific direction and include offset errors (where an instrument doesn't read zero when the quantity is zero) and scale factor errors (where readings consistently differ from true values proportionally) [26] [28].
Comparative Analysis: Type A vs. Type B Uncertainty
Table 1: Fundamental Differences Between Type A and Type B Uncertainty Evaluation
| Characteristic | Type A Uncertainty | Type B Uncertainty |
|---|---|---|
| Basis of Evaluation | Statistical analysis of repeated measurements [25] | Scientific judgment using all available information [27] |
| Probability Distribution | Observed frequency distribution (typically normal) [25] | Assumed based on available knowledge (rectangular, triangular, normal) [27] |
| Data Requirement | Requires multiple repeated measurements | Can be evaluated from single measurements with ancillary information |
| Error Nature | Random errors - unpredictable variations [26] | Systematic errors - consistent, predictable bias [28] |
| Reduction Methods | Increasing sample size, taking repeated measurements [28] | Instrument calibration, procedural controls, randomization [28] |
Uncertainty in Analytical Chemistry: UV Spectroscopy vs. HPLC
Experimental Comparison in Pharmaceutical Analysis
A 2022 study compared UV spectroscopy and High-Performance Liquid Chromatography with Ultraviolet detection (HPLC-UV) for determining piperine content in black pepper, providing direct experimental data on measurement uncertainty between these techniques [8].
Table 2: Performance Comparison of UV Spectroscopy vs. HPLC-UV for Piperine Analysis
| Validation Parameter | UV Spectroscopy | HPLC-UV |
|---|---|---|
| Specificity | Good | Good |
| Linearity | Good | Good |
| Limit of Detection (LOD) | 0.65 | 0.23 |
| Limit of Quantification (LOQ) | Not specified | Not specified |
| Accuracy Range | 96.7 - 101.5% | 98.2 - 100.6% |
| Precision Range (RSD%) | 0.59 - 2.12% | 0.83 - 1.58% |
| Measurement Uncertainty | 4.29% at 49.481 g/kg (k=2) | 2.47% at 34.819 g/kg (k=2) |
The measurement uncertainty in this study was evaluated by developing a mathematical model and combining all uncertainties from selected sources according to NIST guidelines and the Eurachem Guide. Both Type A and Type B standard uncertainties were calculated, with Type A obtained statistically and Type B from given expanded uncertainties [8].
Detailed Methodologies for Uncertainty Assessment
UV Spectroscopy Methodology:
- Sample Preparation: Black pepper samples were ground using a blender and sieved through a 60-mesh screen [8]
- Specificity Assessment: Evaluated by comparing spectra of blank, standard, and black pepper extracts for interference
- Linearity Assessment: Calibration curve constructed from standard solutions analyzed in triplicate
- Precision Evaluation: Repeatability (intra-day precision) determined from five replicated injections of freshly prepared solutions; reproducibility (inter-day precision) assessed by analyzing newly prepared solutions on three consecutive days [8]
HPLC-UV Methodology:
- Chromatographic Conditions: Utilized HPLC with UV detection; mobile phase composition optimized for peak parameters (tailing, symmetry), analysis time, and cost [8] [29]
- Specificity Assessment: Compared retention times of sample peaks with reference standards; evaluated chromatograms for interference [8]
- Linearity: Assessed using linear regression analysis of serial standard solutions prepared in triplicate [8]
- Precision Measurement: Similar to UV methodology with replicated injections under consistent conditions [8]
Uncertainty Propagation in HPLC-UV Systems
Research on uncertainty propagation in HPLC-UV systems identifies key contributors to overall measurement uncertainty. A cause-and-effect analysis reveals that uncertainty in measured HPLC peaks arises from multiple sources [5]:
- Injection Step: Uncertainty in delivered amount from random uncertainty of injected volume and concentration
- Separation Process: Uncertainties from distribution of separated eluate, injected analyte concentration, retention volume, number of theoretical plates, temperature, and flow rate
- Detection Process: Uncertainty in absorbance measurement added to uncertainty in concentration arriving at the detector [5]
Simulation studies indicate that the uncertainty in injection volume typically contributes the greatest uncertainty to the final result in HPLC-UV systems. Additionally, the relative uncertainty of a measured peak height tends to be greater than that of a peak area [5].
Recent Comparative Studies
A 2025 study compared benchtop NMR with HPLC-UV for quantifying methamphetamine hydrochloride, providing additional insights into HPLC-UV performance [30]. While HPLC-UV maintained greater precision (RMSE of 1.1 vs. 2.1 for NMR with quantum mechanical modeling), the study highlighted that NMR permits quantification of all species and potentially enables simultaneous identification and quantification [30].
Another study comparing HPLC and UV spectrophotometric methods for quantification of favipiravir in pharmaceutical formulations further confirmed that the liquid chromatographic method offers higher sensitivity and accuracy compared to spectrophotometric methods, though the latter provides simplicity as it requires "no reagent, pH adjustment or extraction technique" [29].
Visualization of Uncertainty Concepts
Uncertainty Classification and Evaluation Workflow
Uncertainty Evaluation Workflow
Uncertainty Propagation in HPLC-UV Analysis
HPLC-UV Uncertainty Propagation
Essential Research Reagent Solutions
Table 3: Key Reagents and Materials for Uncertainty Comparison Studies
| Reagent/Material | Function in Analysis | Example Specifications |
|---|---|---|
| HPLC-Grade Solvents (Acetonitrile, Methanol) | Mobile phase components; sample preparation | HPLC-grade, low UV cutoff, filtered through 0.22 μm membrane [8] [29] |
| Buffer Salts (Sodium acetate, etc.) | Mobile phase modification; pH control | Analytical grade, dissolved in deionized water [29] |
| Reference Standards | Calibration and quantification | High-purity certified reference materials [8] [29] |
| Column Stationary Phases (C18, etc.) | Compound separation | Specific dimensions (e.g., 4.6 mm × 250 mm, 5.0 μm particle size) [29] |
| Filtration Materials | Mobile phase and sample purification | 0.22 μm or 0.45 μm membrane filters [8] [29] |
| Deionized Water | Solvent for mobile phases and standards | High-purity (e.g., 18.2 MΩ·cm) from water purification systems [8] [29] |
The classification of uncertainty into Type A (random) and Type B (systematic) provides a systematic framework for evaluating measurement reliability in analytical chemistry. Experimental comparisons between UV spectroscopy and HPLC-UV consistently demonstrate that HPLC-UV offers superior sensitivity, accuracy, and lower measurement uncertainty, making it more suitable for applications requiring high precision. However, UV spectroscopy remains valuable for its simplicity, speed, and cost-effectiveness, particularly for routine analyses where its higher uncertainty is acceptable. Understanding and properly evaluating both Type A and Type B uncertainties enables researchers to make informed decisions about method selection and interpret results with appropriate confidence, ultimately enhancing the reliability of analytical measurements in pharmaceutical development and quality control.
Practical Implementation and Pharmaceutical Application Scenarios
In the realm of analytical chemistry, the selection of an appropriate technique is fundamental to obtaining reliable and meaningful data. Ultraviolet (UV) spectroscopy and High-Performance Liquid Chromatography (HPLC) are two cornerstone methods for the quantification of chemical substances. UV spectroscopy measures the absorption of ultraviolet light by a sample, providing a simple and rapid means of quantification for compounds containing chromophores. HPLC, particularly when coupled with a UV detector (HPLC-UV), separates the components of a mixture within a column before detecting them, offering superior specificity for complex samples. This guide provides an objective comparison of their performance, with a specific focus on measurement uncertainty, to assist researchers, scientists, and drug development professionals in selecting the optimal technique for their analytical problem.
Comparative Performance and Measurement Uncertainty
The choice between UV spectroscopy and HPLC is often dictated by the required sensitivity, specificity, and the inherent measurement uncertainty of the technique in a given context. Measurement uncertainty defines the range of values within which the true value of a measurement is expected to lie and is a critical metric for evaluating analytical method performance.
Quantitative Comparison of Validation Parameters
The following table summarizes key validation parameters from direct comparison studies of UV and HPLC methods for specific compounds, illustrating typical performance differences.
Table 1: Comparison of UV and HPLC Method Validation Parameters from Scientific Studies
| Analyte (Matrix) | Method | Linearity (R²) | LOD & LOQ | Precision (% RSD) | Accuracy (% Recovery) | Measurement Uncertainty | Source |
|---|---|---|---|---|---|---|---|
| Repaglinide (Tablets) | UV | >0.999 | 5-30 μg/mL (Range) | <1.50% | 99.63-100.45% | Not specified | [1] |
| HPLC | >0.999 | 5-50 μg/mL (Range) | <1.50% (More precise) | 99.71-100.25% | Not specified | [1] | |
| Piperine (Black Pepper) | UV | Good | LOD: 0.65 μg/mL | 0.59-2.12% | 96.7-101.5% | 4.29% (at 49.48 g/kg) | [8] |
| HPLC-UV | Good | LOD: 0.23 μg/mL | 0.83-1.58% | 98.2-100.6% | 2.47% (at 34.82 g/kg) | [8] | |
| Levofloxacin (Scaffolds) | UV | 0.9999 | 0.05-300 μg/mL (Range) | Not specified | 96.0-99.5% | Not specified | [18] |
| HPLC | 0.9991 | 0.05-300 μg/mL (Range) | Not specified | 96.37-110.96%* | Not specified | [18] |
Note the recovery rate of 110.96% for a medium concentration in the Levofloxacin study highlights the potential for inaccuracy with HPLC in complex matrices when not fully optimized, though it often provides superior accuracy [18].
Analysis of Comparative Data
- Sensitivity and Specificity: HPLC consistently demonstrates lower Limits of Detection (LOD) and Quantification (LOQ) compared to UV spectroscopy, as seen in the piperine study [8]. Its separation step prior to detection allows it to distinguish the analyte from interfering matrix components, which is a significant advantage for complex samples like biological fluids, foods, and novel drug delivery systems [18] [22].
- Precision and Accuracy: Both methods are capable of high precision (%RSD <2%) and excellent accuracy (recoveries close to 100%) [1]. However, HPLC often achieves marginally better precision and is less susceptible to inaccuracies from spectral interferences.
- Measurement Uncertainty: A direct comparison for piperine analysis found that the HPLC-UV method had a significantly lower expanded measurement uncertainty (2.47%) compared to the UV method (4.29%) [8]. This makes HPLC the more reliable choice for applications requiring high metrological rigor.
Detailed Experimental Protocols
To illustrate how these methods are developed and validated, here are the detailed protocols from two key studies.
Protocol 1: Determination of Repaglinide in Tablets
Table 2: Key Research Reagent Solutions for Repaglinide Analysis
| Reagent/Solution | Function in the Analysis |
|---|---|
| Repaglinide Reference Standard | Serves as the primary standard for calibration and quantification. |
| Methanol (HPLC grade) | Acts as the solvent for preparing standard and sample solutions. |
| Orthophosphoric Acid | Used to adjust the pH of the mobile phase to 3.5, optimizing chromatographic separation. |
| Mobile Phase: Methanol:Water (80:20, v/v, pH 3.5) | The liquid medium that carries the sample through the HPLC column, facilitating separation. |
A. UV Spectrophotometric Method [1]
- Standard Solution: A stock solution of repaglinide (1000 μg/mL) was prepared in methanol. Working standard solutions in the range of 5–30 μg/mL were prepared by dilution.
- Sample Preparation: Twenty tablets were weighed and powdered. A portion equivalent to 10 mg of repaglinide was dissolved in methanol, sonicated for 15 minutes, diluted to volume, and filtered. The filtrate was further diluted to a concentration within the linear range.
- Measurement: The absorbance of the standard and sample solutions was measured at 241 nm against a methanol blank.
B. HPLC-UV Method [1]
- Chromatographic Conditions:
- Column: Agilent TC-C18 (250 mm × 4.6 mm, 5 μm)
- Mobile Phase: Methanol and water (80:20, v/v, pH adjusted to 3.5 with orthophosphoric acid)
- Flow Rate: 1.0 mL/min
- Detection Wavelength: 241 nm
- Injection Volume: 20 μL
- Standard and Sample Preparation: Prepared similarly to the UV method, but final dilutions were made with the mobile phase.
Protocol 2: Determination of Piperine in Black Pepper
Table 3: Key Research Reagent Solutions for Piperine Analysis
| Reagent/Solution | Function in the Analysis |
|---|---|
| Piperine Standard | The target analyte used for creating the calibration curve. |
| Methanol & Acetonitrile (HPLC grade) | Used for extraction and as components of the mobile phase. |
| Citric Acid | An additive in the mobile phase to improve chromatographic peak shape. |
| HVLP Filters (0.45 μm) | For removing particulate matter from samples before injection into the HPLC system. |
A. UV Spectrophotometric Method [8]
- Sample Preparation: Black pepper samples were ground and sieved. Piperine was extracted from the powder, and the solution was filtered.
- Measurement: The absorbance of the filtered extract was measured at the λ_max of piperine. The concentration was determined using a pre-established calibration curve.
B. HPLC-UV Method [8]
- Chromatographic Conditions:
- Detection Wavelength: Specific wavelength for piperine (not explicitly stated, typically ~340 nm).
- Calibration: Matrix-matched calibration curves (0.8–200 mg/kg) were used to account for matrix effects.
- Validation: The method was validated for specificity, LOD, LOQ, precision, and accuracy. Measurement uncertainty was estimated by combining all uncertainty sources, such as those from sample preparation and the calibration curve.
Analytical Method Selection Workflow
The following diagram outlines a logical decision-making workflow for selecting between UV spectroscopy and HPLC based on the analytical problem's requirements.
The selection between UV spectroscopy and HPLC is a trade-off between simplicity and analytical power. UV spectroscopy is a robust, cost-effective, and rapid solution for the quantitative analysis of pure substances or simple mixtures where spectral interferences are absent. For complex matrices, such as plant extracts, biological fluids, or formulated drug delivery systems, HPLC-UV is the unequivocally superior technique due to its high specificity, superior sensitivity, and lower measurement uncertainty. The data and protocols presented in this guide provide a scientific foundation for researchers to make an informed, problem-driven choice between these two fundamental analytical techniques.
In the pharmaceutical industry, the choice of analytical technique is crucial for ensuring drug quality, safety, and efficacy. Ultraviolet (UV) spectroscopy and High-Performance Liquid Chromatography (HPLC) are both foundational techniques for quantitative analysis. This guide provides an objective comparison of their performance in three key applications—raw material identification, dissolution testing, and single-component analysis—framed within the critical context of measurement uncertainty.
The core difference between these techniques lies in their operation: UV spectroscopy analyzes a sample's total absorption without separation, whereas HPLC first separates the mixture components before UV detection. This fundamental distinction drives their performance characteristics [31].
| Feature | UV Spectroscopy | HPLC with UV Detection |
|---|---|---|
| Basic Principle | Measures absorption of UV light by a sample without separation [32]. | Separates components via a column, then detects them with a UV detector [1]. |
| Key Application: Single-Component Assay | Direct analysis possible if no interferents present [33]. | Analysis possible even in complex mixtures due to initial separation [1]. |
| Key Application: Dissolution Testing | Ideal for continuous, real-time monitoring via fiber optic probes [34]. | Typically used for offline analysis of collected samples; provides specificity [34]. |
| Key Application: Raw Material ID | Provides a molecular fingerprint for identification via libraries [35]. | Not typically used as a primary method for raw material identification. |
| Sensitivity | Lower sensitivity due to design focused on spectral constraints and larger cell volumes [31]. | Higher sensitivity; detector optimized for small cell volumes and high light throughput [31]. |
| Specificity | Lower; susceptible to interference from other absorbing substances [8]. | High; components are separated before detection, minimizing interference [1] [8]. |
| Analysis Speed | Very fast; minimal sample preparation [1]. | Slower; requires time for chromatographic separation [1]. |
| Cost & Operation | Lower cost, simpler operation [34]. | Higher cost, requires skilled operation and solvent management [34]. |
In-Depth Application Analysis and Experimental Protocols
Single-Component Analysis
This application involves determining the concentration of a single active pharmaceutical ingredient (API), free from interfering absorbers.
UV Spectroscopy Workflow: For a single-component assay, UV spectroscopy relies on direct measurement based on the Beer-Lambert law. The common methodologies are:
- Calibration Curve Method: The most robust approach. Prepare a series of standard solutions of known concentrations, measure their absorbances, and plot absorbance versus concentration to create a calibration curve. The concentration of the unknown sample is determined from its absorbance using this curve [33].
- Standard Absorptivity Method: Uses a previously determined standard absorptivity value (A1cm1%) to calculate concentration directly from the sample's absorbance. This is often used for stable substances with broad absorption bands or when the reference substance is expensive [33].
- Single/Double-Point Standardization: The absorbance of the sample and a single standard solution of the reference substance are measured. The sample concentration is calculated from the proportional relationship: Ctest = (Atest × Cstd) / Astd [33].
HPLC-UV Workflow: The general HPLC method for a single-component assay in a formulation involves:
- Sample Preparation: Powder tablets and extract the API with a suitable solvent like methanol [1].
- Chromatographic Separation: Inject the sample onto a reversed-phase C18 column. A mobile phase, such as methanol and water (80:20 v/v, pH adjusted to 3.5), is pumped at a defined flow rate (e.g., 1.0 mL/min) [1].
- Detection & Quantification: Detect the eluting API peak at its λmax (e.g., 241 nm for repaglinide) and use a calibration curve of peak area versus concentration for quantification [1].
Figure 1: UV Spectroscopy single-component analysis workflow.
Dissolution Testing
Dissolution testing measures the rate and extent at which an API is released from its solid dosage form into a dissolution medium.
UV Spectroscopy Workflow:
- Setup: Use a USP dissolution apparatus (e.g., Apparatus 1 or 2) with the vessel maintained at 37°C [34].
- Sampling & Analysis: Withdraw aliquots from the dissolution vessel at predetermined time points. Filter the samples to remove undissolved particles.
- Measurement: Measure the absorbance of the filtered aliquot, either offline in a cuvette or using an online flow cell. The concentration is calculated using a pre-established calibration curve [34].
- Advanced UV Imaging: UV Surface Dissolution Imaging (SDI) is an advanced technique where a compacted sample is mounted in a flow cell. UV light is used to image the concentration gradient of the drug at the solid-liquid interface in real-time, providing insights into dissolution mechanisms [34] [36].
HPLC-UV Workflow:
- Setup & Sampling: The initial steps are identical—set up the dissolution apparatus and withdraw aliquots at specific times [34].
- Sample Preparation: Filter the aliquots and often perform a dilution step to ensure the concentration falls within the linear range of the HPLC method.
- Chromatographic Analysis: Inject the prepared sample into the HPLC system. The separation and quantification steps follow the same principles as in single-component analysis, providing high specificity [34].
Figure 2: General dissolution testing workflow for UV and HPLC.
Raw Material Identification
This is a quality control step to verify the identity of an incoming raw material, such as an Active Pharmaceutical Ingredient (API) or excipient.
UV Spectroscopy Workflow: While a simple spectrum can be used for identification, Raman spectroscopy is the more recognized and powerful technique for this application.
- Measurement: Place the raw material in a suitable container or use a handheld Raman spectrometer to directly measure the sample [35].
- Spectral Matching: The instrument collects a Raman spectrum, which serves as a unique molecular fingerprint. This spectrum is automatically compared against a pre-established library of reference spectra [35].
- Result: The material is confirmed to pass identification if its spectrum matches a library entry within a predefined confidence threshold [35].
Quantitative Performance Comparison: Measurement Uncertainty
A direct comparison of validation parameters from studies on repaglinide tablets and piperine in black pepper highlights the performance differences, particularly in measurement uncertainty [1] [8].
Table 2: Validation data for the analysis of repaglinide in tablets.
| Parameter | UV Spectroscopy | HPLC-UV |
|---|---|---|
| Linearity (R²) | > 0.999 (5-30 μg/mL) | > 0.999 (5-50 μg/mL) |
| Accuracy (% Recovery) | 99.63 - 100.45% | 99.71 - 100.25% |
| Precision (% RSD) | < 1.50% | < 1.50% |
| Limit of Detection (LOD) | Not specified | Not specified |
Data sourced from [1]
Table 3: Validation data and uncertainty for the analysis of piperine in black pepper.
| Parameter | UV Spectroscopy | HPLC-UV |
|---|---|---|
| Linearity (R²) | Good | Good |
| Accuracy (% Recovery) | 96.7 - 101.5% | 98.2 - 100.6% |
| Precision (% RSD) | 0.59 - 2.12% | 0.83 - 1.58% |
| Limit of Detection (LOD) | 0.65 mg/kg | 0.23 mg/kg |
| Measurement Uncertainty (k=2) | 4.29% (at 49.48 g/kg) | 2.47% (at 34.82 g/kg) |
Data sourced from [8]
The data shows that while both methods can be highly accurate and precise, HPLC-UV generally offers superior sensitivity (lower LOD) and lower measurement uncertainty [8]. The higher uncertainty in UV spectroscopy is often attributed to its lack of separation, making it more susceptible to interference from other sample components that contribute to the total absorbance signal.
Essential Research Reagent Solutions
Table 4: Key reagents and materials used in UV and HPLC methods.
| Item | Function | Example Use |
|---|---|---|
| Methanol / Acetonitrile (HPLC Grade) | Common solvent for preparing standard and sample solutions; mobile phase component [1] [8]. | Extracting repaglinide from tablets [1]. |
| Standard Reference Compound | Provides the known benchmark for qualitative and quantitative analysis [1] [8]. | Creating a calibration curve for piperine quantification [8]. |
| Orthophosphoric Acid | Used to adjust the pH of the mobile phase to improve chromatographic separation (peak shape and retention) [1]. | Mobile phase pH adjustment to 3.5 for repaglinide assay [1]. |
| Ultra-pure Water | Used for preparation of aqueous mobile phases and sample solutions to avoid UV-absorbing contaminants [8]. | Mobile phase component in HPLC analysis of piperine [8]. |
| HVLP Filters (0.45 µm) | For removing particulate matter from samples before injection into the HPLC system or UV spectrometer flow cell [8]. | Filtering black pepper extract prior to HPLC analysis [8]. |
The choice between UV spectroscopy and HPLC is not a matter of one being universally better than the other, but rather selecting the right tool for the specific analytical need.
- UV Spectroscopy is the preferred choice for raw material identification (via Raman) and dissolution testing when speed, cost, and real-time monitoring are priorities, and when the analyte can be measured without interference.
- HPLC-UV is the unequivocal choice for single-component assays in complex matrices and for dissolution testing when maximum specificity and accuracy are required. It provides lower measurement uncertainty and higher sensitivity, making it indispensable for method validation and regulatory filing.
Understanding the capabilities and limitations of each technique allows scientists to make informed decisions that ensure drug quality and patient safety throughout the pharmaceutical development lifecycle.
High-Performance Liquid Chromatography (HPLC) is a cornerstone of modern pharmaceutical analysis, essential for ensuring drug safety and efficacy. This guide examines the performance of HPLC in key application areas, with a specific focus on its measurement reliability compared to UV spectroscopy, providing supporting experimental data for informed analytical decision-making.
HPLC in Pharmaceutical Analysis: Core Applications
HPLC plays a critical role throughout the drug development and quality control lifecycle. Its applications ensure that pharmaceutical products are pure, stable, and consistently manufactured to the highest standards.
Impurity Profiling: Identifying and quantifying known and unknown impurities in active pharmaceutical ingredients (APIs) and finished products is crucial for patient safety. A stability-indicating method for a triple-combination dry powder inhaler (Budesonide, Glycopyrronium, and Formoterol Fumarate) demonstrates this capability. The method successfully separated and quantified nine known toxic impurities in a single run, with low limits of detection (LOD) and quantification (LOQ), ensuring the product's safety profile [37]. Similarly, a robust method was developed for Fosamprenavir, an antiretroviral drug, effectively separating it from five potential process-related impurities (Isomer, Amino, Propyl, Nitro, and Amprenavir) within a 10-minute runtime, supporting rigorous quality monitoring [38].
Stability Studies: These studies assess how the quality of a drug substance or product varies with time under environmental factors. The aforementioned method for the triple-combination inhaler was validated as stability-indicating, meaning it can accurately quantify the active ingredients and detect degradation products that might form under stress conditions, providing a direct assessment of drug shelf-life [37].
Analysis of Multicomponent Formulations: HPLC efficiently analyzes products containing multiple active ingredients. A method for a powder containing Paracetamol, Phenylephrine hydrochloride, and Pheniramine maleate was optimized for both the assay of active ingredients and the quantification of a key impurity, 4-aminophenol. This highlights HPLC's versatility in handling complex mixtures for different quality attributes [39]. Furthermore, a rapid method was developed for the simultaneous determination of five COVID-19 antiviral drugs (Favipiravir, Molnupiravir, Nirmatrelvir, Remdesivir, and Ritonavir) in pharmaceutical formulations, showcasing its power in analyzing complex therapeutic regimens [40].
Performance Comparison: HPLC-UV vs. UV Spectroscopy
The choice of analytical technique is often a balance between performance, cost, and complexity. A direct comparison of HPLC-UV and UV Spectroscopy for the determination of piperine in black pepper provides valuable experimental data on their performance and measurement uncertainty [8].
Table 1: Performance Comparison of UV Spectroscopy and HPLC-UV for Piperine Analysis [8]
| Performance Parameter | UV Spectroscopy | HPLC-UV |
|---|---|---|
| Specificity | Good | Good |
| Linearity | Good | Good |
| Limit of Detection (LOD) | 0.65 | 0.23 |
| Accuracy (%) | 96.7 – 101.5 | 98.2 – 100.6 |
| Precision (% RSD) | 0.59 – 2.12 | 0.83 – 1.58 |
| Measurement Uncertainty (k=2) | 4.29% (for 49.481 g/kg) | 2.47% (for 34.819 g/kg) |
Comparative Analysis and Uncertainty
The data shows that while both methods performed well for specificity and linearity, HPLC-UV demonstrated superior sensitivity, with a lower LOD. The accuracy and precision for both techniques were within acceptable limits. A key differentiator is the measurement uncertainty: HPLC-UV showed significantly lower uncertainty (2.47%) compared to UV spectroscopy (4.29%). This indicates that HPLC-UV provides a more reliable and precise measurement, which is critical for making confident decisions in quality control and regulatory compliance [8].
Detailed Experimental Protocols
- Objective: To develop and validate a single-run RP-HPLC method for quantifying nine known impurities in Budesonide, Glycopyrronium, and Formoterol Fumarate combination products.
- Chromatographic Conditions:
- Column: Bakerbond C18, 5 µm, 250 × 4.6 mm
- Mobile Phase: Gradient with (A) pH-2 buffer (KH₂PO₄ and sodium 1-octanesulfonate) and (B) acetonitrile:methanol (90:10 %v/v)
- Flow Rate: 0.8 mL/min
- Temperature: 35°C
- Detection: 220 nm for Glycopyrronium/Formoterol impurities; 240 nm for Budesonide impurities
- Injection Volume: Not specified
- Sample Preparation: The drug product was dissolved in a diluent of methanol and water (50:50 %v/v).
- Validation Outcomes: The method was validated per ICH Q2(R2). It was found to be specific, precise (%RSD 2.95–11.31), accurate (recovery 90.9–113.8%), and linear (r > 0.97) for all impurities and active drugs.
- Objective: To develop a rapid, single-method RP-HPLC approach for the simultaneous quantification of five COVID-19 antivirals.
- Chromatographic Conditions:
- Column: Hypersil BDS C18 (150 mm × 4.6 mm; 5 µm)
- Mobile Phase: Isocratic, water:methanol (30:70 v/v, pH 3.0 adjusted with 0.1% ortho-phosphoric acid)
- Flow Rate: 1.0 mL/min
- Temperature: 25°C
- Detection: 230 nm
- Injection Volume: 20 µL
- Run Time: 6 minutes
- Sample Preparation: Stock solutions (1000 µg/mL) of each drug were prepared in methanol and then diluted to working concentrations with methanol.
- Validation Outcomes: The method was linear (r² ≥ 0.9997) over 10–50 µg/mL, precise (RSD < 1.1%), and accurate (trueness 99.59-100.08%). It was successfully applied to pharmaceutical formulations.
Workflow and Technical Considerations
The following workflow outlines a structured approach for developing and validating an HPLC method, incorporating uncertainty assessment as a critical final step.
(Diagram: A generalized workflow for HPLC method lifecycle, highlighting the role of validation in uncertainty assessment.)
The Scientist's Toolkit: Essential Reagents and Materials
Table 2: Key Reagents and Materials for HPLC Analysis
| Item | Function / Role | Example from Literature |
|---|---|---|
| C18 Chromatographic Column | The stationary phase for reverse-phase separation; a core component. | Bakerbond C18 [37], Hypersil BDS C18 [40], Zorbax SB-Aq [39] |
| HPLC-Grade Solvents | Mobile phase components; high purity is critical to minimize background noise. | Acetonitrile, Methanol [37] [40] [41] |
| Buffer Salts & pH Modifiers | Control the pH and ionic strength of the mobile phase, critical for peak shape and separation. | Potassium dihydrogen phosphate [37], Ortho-phosphoric acid [40] [41], Ammonium formate [42] |
| Reference Standards | Highly characterized materials used to calibrate the system and quantify analytes. | USP reference standards [37], Qualified in-house reference materials [42] |
| Syringe Filters | Clarify samples by removing particulate matter before injection onto the column. | 0.45 µm nylon filters [37] [42] |
HPLC remains the dominant technique for impurity profiling, stability studies, and analysis of complex formulations due to its superior selectivity, sensitivity, and ability to separate multiple components. The comparative data clearly shows that HPLC-UV offers a significant advantage over UV spectroscopy in terms of measurement uncertainty, providing a more reliable foundation for critical quality decisions. For laboratories where the highest level of measurement certainty is required—especially for regulatory submissions and quality control—HPLC is the unequivocal choice.
In the pharmaceutical industry, ensuring drug quality and safety necessitates robust analytical methods for the quantification of active ingredients. UV-Spectrophotometry and High-Performance Liquid Chromatography (HPLC) are two cornerstone techniques used for this purpose. This case study provides a direct comparison of these two techniques for the simultaneous analysis of Cefixime Trihydrate (CEFI), a third-generation cephalosporin antibiotic, and Moxifloxacin Hydrochloride (MOXI), a fourth-generation fluoroquinolone antibiotic [43] [44]. The combination of these two drugs is used to treat respiratory tract infections, and robust analytical methods are required for routine quality control and regulatory compliance [44]. The broader thesis context of this comparison is to evaluate the measurement uncertainty associated with each technique, a critical factor in making reliable pharmaceutical equivalence decisions [45].
Experimental Protocols & Workflows
UV-Spectrophotometry Methods
For the simultaneous estimation of CEFI and MOXI in a combination formulation, two UV-spectrophotometric methods were developed and validated [43] [44].
- Absorbance Ratio Method (Q-Absorption): This method utilizes the measurement of absorbance at two wavelengths: an iso-absorptive point and the λ-max of one of the drugs. The linearity for both CEFI and MOXI was established in the concentration range of 3–15 μg/ml [44].
- First-Order Derivative Spectroscopy: This technique involves converting the normal zero-order spectra into first-order derivative spectra. The concentrations of CEFI and MOXI were determined at wavelengths where the derivative value of the other drug was zero, thus eliminating interference [43].
The sample preparation for both UV methods involved dissolving appropriate amounts of the synthetic mixture or marketed formulation in a suitable solvent, followed by dilution to the desired concentration within the linear range [44].
RP-HPLC Method
A reversed-phase high-performance liquid chromatography (RP-HPLC) method was developed to separate and quantify CEFI and MOXI simultaneously [43] [44].
- Chromatographic Conditions: The analysis was performed using a C18 column (250 mm × 4.6 mm, 5 μm). The mobile phase consisted of a mixture of 20 mM potassium dihydrogen phosphate buffer (pH adjusted to 3.0) and acetonitrile in a gradient elution mode. The flow rate was maintained at 1.0 ml/min, and the detection was carried out using a UV detector set at 295 nm [44].
- Sample Analysis: Under these optimized conditions, CEFI and MOXI were well-separated with retention times of approximately 2.24 minutes and 3.16 minutes, respectively, with a total run time of 5 minutes [44]. The method demonstrated excellent resolution, precision, and robustness.
The following workflow diagram illustrates the key stages of both analytical processes and their role in quality assessment, highlighting the additional separation step in HPLC that contributes to its higher specificity.
Critical Reagents and Research Solutions
The successful execution of analytical methods relies on specific, high-quality reagents and materials. The following table details key components used in the featured experiments for the analysis of CEFI and MOXI.
| Research Reagent/Material | Function & Application in Analysis |
|---|---|
| C18 Chromatographic Column | The stationary phase for RP-HPLC; enables separation of CEFI and MOXI based on hydrophobicity [43] [44]. |
| Potassium Dihydrogen Phosphate Buffer | A component of the HPLC mobile phase; helps control pH to optimize separation and peak shape [44]. |
| HPLC-Grade Acetonitrile | An organic modifier in the HPLC mobile phase; facilitates elution of analytes from the column [44]. |
| 1,10-o-Phenanthroline / 2,2'-Bipyridyl | Complexing agents used in specific UV-spectrophotometric methods for MOXI to form colored complexes for measurement [46]. |
| Methanol | A solvent used for dissolving samples and standards in both UV and HPLC method development [44]. |
| Formic Acid | Used in mobile phase to improve chromatographic peak shape and as a catalyst in derivatization reactions [47] [48]. |
Comparative Performance Data
The developed UV and HPLC methods were rigorously validated as per International Conference on Harmonisation (ICH) guidelines. The following table summarizes key validation parameters, providing a direct comparison of the performance of both techniques for the simultaneous assay [43] [44].
Table 1: Summary of validation parameters for UV-Spectrophotometry and RP-HPLC methods for CEFI and MOXI analysis.
| Validation Parameter | UV-Spectrophotometry (Q-Absorption) | UV-Spectrophotometry (First-Order Derivative) | RP-HPLC |
|---|---|---|---|
| Linearity Range | 3–15 μg/mL for both drugs [44] | 3–15 μg/mL for both drugs [44] | 5–25 μg/mL for both drugs [44] |
| Precision (% R.S.D.) | ≤ 1.5% [44] | ≤ 1.5% [44] | ≤ 2.0% [44] |
| Accuracy (% Recovery) | 99.3% - 100.4% [44] | 99.5% - 100.6% [44] | 99.1% - 101.2% [44] |
| Assay of Marketed Formulation | CEFI: 99.59%, MOXI: 98.84% [44] | Data specific to formulation not provided [43] | CEFI: 99.59%, MOXI: 98.84% [44] |
| Limit of Detection (LOD) | Higher | Higher | Lower (approx. 0.30 μg/mL for CEFI, 0.132 μg/mL for MOXI derivative) [48] |
| Specificity | Lower (relies on mathematical processing for separation) | Lower (relies on mathematical processing for separation) | Higher (physical separation of components) [43] |
Measurement Uncertainty and Risk Assessment
Measurement uncertainty is an omnipresent factor in analytical chemistry that must be quantified to make reliable conformity decisions [5] [45]. In HPLC with UV detection, uncertainty originates from multiple steps, including injection volume, sample concentration, flow rate, and detection [5]. One study identified that uncertainty in injection volume contributes the greatest uncertainty to the final result in HPLC [5]. For UV-spectrophotometry, uncertainty can arise from factors like preparation of standards, instrumental noise, and matrix effects.
This uncertainty directly impacts pharmaceutical quality control. When assessing pharmaceutical equivalence—demonstrating that a generic drug is equivalent to a reference drug—measurement uncertainty can lead to false decisions [45]. The combined total risk of falsely accepting a non-equivalent product or falsely rejecting an equivalent one can be estimated using the uncertainty of the analytical method [45]. HPLC methods, with their superior specificity and ability to separate drugs from their degradation products, can reduce this risk by providing more accurate and precise data, especially for drugs like cefixime which is known to degrade and form impurities [47].
The choice between UV-Spectrophotometry and RP-HPLC for the simultaneous analysis of Cefixime and Moxifloxacin depends on the specific requirements of the analysis, balancing simplicity and cost against specificity and robustness.
- UV-Spectrophotometry offers simplicity, rapidity, and cost-effectiveness. The Q-Absorption and First-Order Derivative methods are well-suited for the routine analysis of formulations where the two drugs are the only major absorbing components and no significant interference from excipients is present [43]. However, it has higher detection limits and lower specificity, making it more susceptible to uncertainty from matrix effects.
- RP-HPLC is the more powerful and reliable technique. It provides high resolution, specificity, sensitivity, and the ability to detect and quantify degradation products and impurities, as demonstrated for cefixime [47]. This leads to lower measurement uncertainty and, consequently, a lower risk of false equivalence decisions [45]. It is the unequivocal choice for stability studies, method transfer, and situations requiring the highest level of data integrity.
In conclusion, while UV-spectrophotometry serves as a good screening and rapid analysis tool, RP-HPLC is the definitive technique for the precise, accurate, and robust simultaneous quantification of Cefixime and Moxifloxacin in pharmaceutical formulations, particularly when the impact of measurement uncertainty on critical decisions is a primary concern.
In the realm of pharmaceutical development, dissolution testing serves as a critical bridge between drug production and therapeutic effectiveness, enabling scientists to predict in-vivo performance and ensure batch-to-batch consistency [49]. For decades, high-performance liquid chromatography (HPLC) has stood as the gold standard for analyzing dissolution samples, offering high specificity and sensitivity. However, the emergence of advanced imaging techniques, particularly UV dissolution imaging, presents a paradigm shift toward real-time, spatially resolved monitoring of drug release processes.
This guide objectively compares the operational performance, measurement uncertainty, and application potential of UV dissolution imaging against traditional HPLC and UV spectroscopy for dissolution testing. The content is framed within a broader research thesis comparing measurement uncertainty between UV spectroscopy and HPLC, providing drug development professionals with experimental data and protocols to inform their analytical method selection. The comparison reveals that while HPLC remains indispensable for complex mixtures requiring high specificity, UV dissolution imaging offers unparalleled real-time insights into dissolution mechanisms with comparable uncertainty profiles to established techniques.
Technical Principles and Comparative Advantages
Fundamental Operating Principles
UV Dissolution Imaging: This emerging technology utilizes light in the wavelength range of 190-400 nm to provide visualisation of dissolution phenomena at the solid-liquid interface while simultaneously obtaining concentration measurements [36]. It captures spatially resolved drug concentration gradients and transport phenomena in real-time, offering insights into dissolution mechanisms not available through traditional methods.
Traditional UV Spectroscopy: Based on the absorbance of light by drug molecules in solution, this method provides a single absorbance value used to determine concentration through Beer-Lambert law principles [49]. It measures the bulk concentration in dissolution vessels without spatial information about the dissolution process.
HPLC Analysis: This technique separates mixture components using a chromatographic column before quantifying them via UV detection [50]. It offers high specificity by physically separating the analyte from excipients, degradation products, or other interfering substances that may co-elute in direct UV methods.
Performance Comparison
Table 1: Comparative Analysis of Key Performance Parameters
| Parameter | UV Dissolution Imaging | Traditional UV Spectroscopy | HPLC Analysis |
|---|---|---|---|
| Analytical Speed | Real-time monitoring with temporal resolution | Immediate analysis post-sampling; sipper systems enable rapid analysis [49] | Longer run times (typically 10-60 minutes) plus mobile phase preparation [49] |
| Spatial Information | Provides concentration gradients and visualization of dissolution phenomena [36] | Bulk solution measurement only | Bulk solution measurement only |
| Specificity | Moderate; limited by absorbance spectra overlap | Low; susceptible to interference from excipients with similar UV profiles [49] | High; separation eliminates most interference issues [49] |
| Cost Considerations | High initial instrument investment | Significantly lower operational costs; no organic solvents or expensive columns [49] | High operational costs; organic solvents, column maintenance, and disposal [49] |
| Measurement Uncertainty Contributors | Hydrodynamic modeling, image analysis, flow conditions [36] | Primarily quantification step (47% of total uncertainty) [51] | Multiple sources: sampling, dissolution conditions, and quantification [52] |
Measurement Uncertainty Framework
Measurement uncertainty is a fundamental metrological parameter that characterizes the dispersion of values reasonably attributable to a measurand, reflecting the degree of confidence in analytical results [3]. Understanding the distinct uncertainty profiles of each technique is essential for method selection and risk assessment in pharmaceutical analysis.
UV Spectroscopy Uncertainty: Studies of dissolution testing for prednisone tablets revealed that the quantification step contributed 47% of the overall measurement uncertainty, significantly higher than sampling (24%) and dissolution steps (29%) [51]. This highlights that even traditional UV methods face substantial analytical uncertainty alongside sampling variability.
HPLC Uncertainty: Research on glyburide tablets demonstrated different uncertainty distribution, with sampling uncertainty contributing 76% of overall uncertainty, followed by the dissolution step at 22%, while the quantification step contributed only 2% [52]. This suggests HPLC quantification itself provides high precision, with most uncertainty arising from physical processes rather than separation and detection.
UV Imaging Uncertainty: The emerging nature of this technique means comprehensive uncertainty budgets are less established. However, known significant sources include the suitability of hydrodynamic models for prevailing flow conditions and variability in image analysis algorithms [36]. Unlike traditional methods, it also introduces spatial uncertainty components related to resolution limitations.
Uncertainty Evaluation and Management
A four-step process for uncertainty estimation includes: (1) defining the measurand and relationship with input quantities, (2) identifying uncertainty sources, (3) quantifying uncertainty contributions, and (4) calculating combined standard uncertainty and expanded uncertainty with appropriate coverage factor (typically k=2 for 95% confidence) [3].
Cause-and-effect diagrams (Ishikawa diagrams) provide structured approaches to identify uncertainty sources in chromatographic analysis, including factors like column temperature, carrier gas flow-rate, injection parameters, and detector noise [3]. For dissolution testing regardless of analytical finish, sampling uncertainty often represents a significant component that must be evaluated through appropriate experimental designs such as the duplicate method [51].
Diagram 1: Measurement uncertainty sources in dissolution testing. This cause-and-effect diagram illustrates the major contributors to uncertainty across different analytical techniques, highlighting technique-specific factors that must be considered in method validation and uncertainty budgeting.
Experimental Protocols and Methodologies
UV Dissolution Imaging Protocol
Instrumentation and Setup: UV dissolution imaging systems typically consist of a UV light source, flow-through cell with imaging capabilities, and CCD camera detector. The technology has evolved to include USP type IV-like whole dose cells facilitating studies on tablets and capsules [36]. A typical configuration uses quartz flow cells with pathlengths of 1-10 mm, optimized for the concentration range of interest.
Sample Analysis Procedure:
- Place dosage form (single particle to whole tablet) in the flow cell
- Establish continuous flow of dissolution medium (typically physiological pH aqueous solutions)
- Initiate image acquisition at predetermined intervals (e.g., 1-10 seconds)
- Monitor dissolution process via absorbance at appropriate wavelength (e.g., 280 nm for carbamazepine)
- Analyze concentration gradients and dissolution rates using appropriate software algorithms
- Apply hydrodynamic model to convert spatial and temporal data to dissolution rates
Data Processing: The raw imaging data requires processing to convert absorbance values to concentration maps using Beer-Lambert principles. Challenges include accounting for light scattering effects, non-uniform illumination, and establishing appropriate calibration curves for quantitative analysis [36].
Traditional UV Spectroscopy Protocol
Instrumentation: Double-beam spectrophotometer with fixed slit width (typically 1.5 nm) connected to computer-controlled software, using 1 cm quartz cells [50]. Modern systems often incorporate sipper attachments for automated sampling from dissolution vessels.
Sample Analysis Procedure:
- Withdraw aliquots from dissolution vessels at predetermined time points
- Filter samples if necessary (0.45 µm membrane filter)
- Measure absorbance against blank (dissolution medium) at wavelength of maximum absorption
- Calculate concentration using pre-established calibration curve
- Apply non-derivative techniques (absorbance ratio, compensation) or derivative techniques (first-order derivative, ratio spectra first-order derivative) for mixture analysis [50]
Method Validation: Key parameters include linearity range (e.g., 60-140 µg/mL for amoxicillin and cloxacillin), precision (RSD typically <2%), and accuracy through recovery studies [50].
HPLC Analysis Protocol
Instrumentation: HPLC system with diode-array detector, using reversed-phase C18 columns (e.g., 150 × 4.6 mm, 5µm) at ambient temperature [50] [53].
Chromatographic Conditions:
- Mobile Phase: Variable based on analyte; for penicillins: water-based; for organic acids: phosphoric acid/formic acid with ammonium formate [50] [53]
- Flow Rate: Typically 0.5-2.0 mL/min
- Injection Volume: 5-20 µL
- Detection: UV at analyte-specific wavelength (e.g., 260-280 nm)
Sample Preparation:
- Withdraw dissolution medium aliquots
- Filter through 0.45 µm membrane filter
- Dilute if necessary with mobile phase
- Transfer to HPLC vials for analysis
System Suitability: Parameters including column plate counts, peak symmetry, retention time reproducibility, and resolution must meet established criteria before analysis [49].
Comparative Experimental Data
Quantitative Performance Metrics
Table 2: Experimental Performance Data from Comparative Studies
| Study Focus | Analytical Method | Key Performance Metrics | Measurement Uncertainty |
|---|---|---|---|
| Amoxicillin & Cloxacillin in Capsules [50] | HPLC | Linearity: 60-140 µg/mL; Specificity: High (separation achieved) | Interchangeable with UV; statistical comparison showed no significant difference |
| Amoxicillin & Cloxacillin in Capsules [50] | UV Spectrophotometry | Linearity: 60-140 µg/mL; RSD: <2.5%; Accuracy: Comparable to HPLC | Interchangeable with HPLC for routine analysis |
| Prednisone Tablets Dissolution [51] | UV Spectroscopy | Overall Uncertainty: 2.2% (below target 2.5%); Quantification Contribution: 47% of total uncertainty | Sampling: 24%; Dissolution: 29%; Quantification: 47% |
| Glyburide Tablets Dissolution [52] | HPLC | Overall Uncertainty: Not specified; Quantification Contribution: 2% of total uncertainty | Sampling: 76%; Dissolution: 22%; Quantification: 2% |
| Organic Acids in Food [53] | HPLC-DAD | Validation parameters confirmed: Selectivity, Linearity (R²>0.999), LOD: 0.3-2.3 mg/L, LOQ: 1.0-7.0 mg/L | Measurement uncertainty assessed as part of validation |
Application-Specific Performance
Low Solubility Compounds: For BCS Class II drugs like glyburide with low solubility and high permeability, sampling uncertainty dominated the uncertainty budget (76%) regardless of analytical finish, highlighting the importance of dissolution conditions over detection method for such compounds [52].
Complex Matrices: In cosmetic products containing bakuchiol, HPLC provided definitive identification and quantification even in complex oil-based formulations where UV spectroscopy faced challenges with complete dissolution and proper extraction [6].
Formulation Development: UV dissolution imaging has proven particularly valuable for understanding drug-excipient interactions, form selection, and intrinsic dissolution rate determinations, offering insights beyond concentration measurements alone [36].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagent Solutions for Dissolution Testing
| Item | Function/Application | Technical Specifications |
|---|---|---|
| Dissolution Media | Simulate physiological conditions; typically aqueous solutions at physiological pH [49] | Purified water; phosphate buffers (pH 7.3); biorelevant media |
| HPLC Mobile Phase | Solvent system for chromatographic separation | Varies by analyte: water with buffers; acetonitrile with formic acid (1%); phosphoric acid solutions [50] [53] |
| Reference Standards | Method calibration and quantification | Certified reference substances (CRS); high purity (≥95-98%) [50] [52] |
| Membrane Filters | Sample clarification before analysis | 0.45 µm porosity; compatible with dissolution medium [50] |
| UV Flow Cells | Containment for imaging and spectroscopy | Quartz material; pathlengths 1-10 mm; various designs for different dosage forms [36] |
The comparative analysis of UV dissolution imaging, traditional UV spectroscopy, and HPLC for dissolution testing reveals a complex landscape where method selection depends heavily on application requirements, stage of development, and uncertainty tolerance.
UV dissolution imaging represents a significant advancement for understanding dissolution mechanisms through real-time, spatially resolved monitoring, particularly valuable in early development for form selection, intrinsic dissolution rate determination, and drug-excipient compatibility studies [36]. However, its higher variability from hydrodynamic modeling and image analysis requires careful consideration for quality control applications.
Traditional UV spectroscopy offers compelling advantages for routine quality control where speed, cost-effectiveness, and simplicity are prioritized, particularly for formulations without significant interference issues [49]. Its higher proportional uncertainty from the quantification step necessitates robust method validation.
HPLC maintains its position as the reference method for complex matrices, providing superior specificity and lower quantification uncertainty, making it indispensable for method development and validating simpler techniques [50] [53]. The technique's higher operational costs and longer analysis times continue to drive interest in alternative methods.
For researchers and pharmaceutical professionals, the evolving methodology landscape offers multiple pathways for dissolution testing, with UV dissolution imaging emerging as a powerful complementary technique rather than a direct replacement for established methods. Strategic method selection should balance information needs, uncertainty profiles, and operational constraints throughout the drug development lifecycle.
Minimizing Uncertainty: Strategies for Enhanced Accuracy and Precision
In the comparison of measurement uncertainty between UV spectroscopy and High-Performance Liquid Chromatography (HPLC), the proper evaluation of calibration uncertainty is a critical differentiator. A prevalent and significant error in this process is double counting, where the same source of uncertainty is incorporated more than once into the uncertainty budget. This mistake can lead to a substantial overestimation of the combined measurement uncertainty, compromising the reliability of analytical results and misleading conclusions in method comparison studies. This guide examines this pitfall within the context of comparing UV spectroscopy and HPLC, providing structured data and protocols to support robust, error-free uncertainty analysis for researchers and drug development professionals.
Analytical Technique Comparison: UV Spectroscopy vs. HPLC
The fundamental performance differences between UV spectroscopy and HPLC establish the context for evaluating their measurement uncertainties. The following table summarizes a direct comparison of validation parameters for the determination of piperine in black pepper, highlighting key distinctions relevant to uncertainty budgets [8].
| Performance Parameter | UV Spectroscopy | HPLC-UV |
|---|---|---|
| Limit of Detection (LOD) | 0.65 | 0.23 |
| Accuracy Range | 96.7 - 101.5% | 98.2 - 100.6% |
| Precision (RSD Range) | 0.59 - 2.12% | 0.83 - 1.58% |
| Measurement Uncertainty | 4.29% (at 49.481 g/kg) | 2.47% (at 34.819 g/kg) |
| Key Strengths | Good specificity and linearity; rapid analysis [8]. | Greater sensitivity and accuracy; good specificity and linearity [8]. |
As the data indicates, HPLC generally offers superior sensitivity and lower measurement uncertainty under the conditions reported. A correct uncertainty evaluation is essential for making such valid comparisons.
The Perils of Double Counting in Calibration
Double counting occurs primarily when the precision component of uncertainty is counted twice: once as an individual contribution and again within the "whole method" performance data, such as the overall method precision estimate [54].
The Problem with the Common Calibration Formula
A typical formula for the standard uncertainty from a linear calibration curve is often expressed as:
[ u(x{pred}) = \frac{s{y/x}}{b} \sqrt{\frac{1}{m} + \frac{1}{n} + \frac{(x{pred} - \bar{x})^2}{S{xx}}} ]
Where:
- ( s_{y/x} ) is the residual standard deviation
- ( b ) is the slope
- ( m ) is the number of sample replicates
- ( n ) is the number of calibration standards
- ( x_{pred} ) is the predicted concentration
- ( \bar{x} ) is the mean of the calibration concentrations
- ( S_{xx} ) is the sum of squares of the concentration deviations
The mistake lies in using this full formula when the precision component (( s_{y/x}/b )), which captures random variations from the entire analytical procedure, is already included in the overall method precision estimate. Using the full formula alongside a separate method precision component results in double counting the random effects of the measurement process [54].
Consequences for Method Comparison
In the context of comparing UV and HPLC:
- Overstated Uncertainties: Double counting inflates the combined uncertainty of a method, making a precise technique like HPLC appear less reliable than it truly is.
- Invalid Comparisons: An inflated uncertainty for one method can obscure real performance differences, leading to incorrect conclusions about which technique is more fit-for-purpose.
- Inefficient Resource Allocation: Overestimating uncertainty might lead laboratories to invest in unnecessary instrument upgrades or additional replication to meet uncertainty targets.
Detailed Experimental Protocols
To illustrate how uncertainty data is generated for such comparisons, here are the summarized methodologies from the piperine study and a pharmaceutical analysis.
This protocol directly generated the comparison data in Table 1.
- Sample Preparation: Black pepper samples were ground using a blender and sieved through a 60-mesh screen.
- UV Spectroscopy Analysis:
- Extraction: Piperine was extracted from the prepared sample.
- Measurement: The absorbance of the extract was measured at a specific wavelength.
- Quantification: Concentration was determined using a pre-established calibration curve.
- HPLC-UV Analysis:
- Chromatography: Separations were performed using a specified column and a mobile phase of methanol and acetonitrile.
- Detection: UV detection was used to identify and quantify the piperine peak based on its retention time.
- Method Validation: Both methods were validated for parameters including specificity, LOD, LOQ, linearity, accuracy, and precision according to ICH and AOAC guidelines.
- Uncertainty Estimation: Measurement uncertainty was evaluated by combining all uncertainties from selected sources according to the NIST guidelines and the Eurachem Guide.
This protocol exemplifies a common application in drug development.
- HPLC-UV Conditions:
- Column: Reverse-phase C18 column.
- Mobile Phase: Acetonitrile and 0.5% triethylamine (pH adjusted to 7.5).
- Flow Rate: 1.0 mL/min.
- Detection: 222 nm.
- Internal Standard: Ibuprofen.
- Standard Solution Preparation: A stock solution of Dexibuprofen was prepared and diluted to concentrations of 10.0-60.0 μg/mL for the calibration curve.
- Sample Solution Preparation: Twenty tablets were weighed and finely powdered. A portion equivalent to a single dose was dissolved and extracted with the mobile phase, then filtered and diluted.
- Validation: The method was validated for linearity, precision (repeatability and intermediate precision), accuracy (recovery), and specificity.
Visualizing Uncertainty Components and Their Relationships
The following diagram maps the key components of measurement uncertainty for a typical chromatographic or spectroscopic analysis, highlighting the area where double counting most frequently occurs.
Uncertainty Budget Structure and Double Counting Risk: This workflow shows how calibration uncertainty is one of several components contributing to combined measurement uncertainty. The dashed line highlights the critical overlap between calibration precision and overall method precision, which is the primary source of double counting errors.
The Scientist's Toolkit: Essential Reagents and Materials
The following table lists key materials used in the featured experiments, which are also standard for developing and validating UV and HPLC methods in pharmaceutical analysis.
| Item | Function | Example from Protocols |
|---|---|---|
| HPLC-Grade Solvents | Form the mobile phase to carry the sample through the HPLC column; high purity is essential to minimize baseline noise and ghost peaks. | Methanol, Acetonitrile, Water [8] [55]. |
| Analytical Reference Standards | Used to prepare calibration curves for accurate quantification of the target analyte. | Piperine standard [8], Dexibuprofen standard [55]. |
| Chromatography Columns | The heart of the separation system where compounds in the sample are resolved. | C18 column [55] [56]. |
| Solid-Phase Extraction (SPE) Cartridges | Used for complex samples to clean up and pre-concentrate the analyte, improving sensitivity and accuracy. | Activated charcoal cartridges (mentioned in [54] context). |
| Syringe Filters | Remove particulate matter from samples before injection, protecting the instrument and column from damage. | 0.45 µm HVLP filters [8], 0.45 µm membrane filter [55]. |
| Internal Standard | A compound added in a constant amount to samples and standards to correct for variability in injection volume and sample processing. | Ibuprofen (used in Dexibuprofen method) [55]. |
A rigorous and accurate comparison of measurement uncertainty between UV spectroscopy and HPLC is fundamental to selecting the optimal analytical technique. Avoiding the common mistake of double counting in calibration uncertainty is not merely a statistical exercise; it is a necessary practice for obtaining a true picture of method performance. By using the correct formulae, understanding the components of the uncertainty budget, and following detailed, validated protocols, scientists can ensure their uncertainty estimates are both realistic and reliable. This disciplined approach provides a solid foundation for making informed decisions in method development and drug analysis, ultimately supporting the delivery of safe and effective pharmaceuticals.
In the realm of analytical chemistry, the choice of technique is pivotal to obtaining reliable and accurate data. Ultraviolet (UV) spectroscopy and High-Performance Liquid Chromatography (HPLC) are two fundamental methods used for quantitative analysis, each with distinct advantages and limitations. This guide provides a objective comparison of their performance, with a particular focus on measurement uncertainty, a critical parameter for data integrity in research and drug development. The content is framed within a broader thesis comparing the measurement uncertainty of UV spectroscopy versus HPLC, offering experimental data and protocols to guide scientists in method selection and optimization.
Performance Comparison: UV Spectroscopy vs. HPLC
A direct comparison of analytical techniques is essential for selecting the most appropriate method. The following section objectively compares the performance of UV spectroscopy and HPLC based on key validation parameters and measurement uncertainty.
Quantitative Performance Data
The table below summarizes a direct, experimental comparison of UV spectroscopy and HPLC-UV for the determination of piperine in black pepper, highlighting critical validation parameters [8].
Table 1: Comparison of UV and HPLC-UV Methods for Piperine Analysis
| Performance Parameter | UV Spectroscopy | HPLC-UV |
|---|---|---|
| Specificity | Good | Good |
| Linearity | Good | Good |
| Limit of Detection (LOD) | 0.65 mg/kg | 0.23 mg/kg |
| Limit of Quantification (LOQ) | Information Missing | Information Missing |
| Accuracy (Recovery Range) | 96.7 - 101.5% | 98.2 - 100.6% |
| Precision (RSD Range) | 0.59 - 2.12% | 0.83 - 1.58% |
| Measurement Uncertainty | 4.29% (at 49.481 g/kg) | 2.47% (at 34.819 g/kg) |
A separate study analyzing xylitol in foods further supports this trend, where HPLC with a UV detector (after derivatization) showed superior sensitivity with an LOD of 0.01 mg/L compared to other HPLC detectors like evaporative light scattering (ELSD) and refractive index (RID) [57].
Key Findings and Recommendations
- Sensitivity and Accuracy: HPLC demonstrates lower LOD and a tighter accuracy range, making it more suitable for quantifying analytes at trace levels or requiring high accuracy [8].
- Measurement Uncertainty: HPLC exhibits lower measurement uncertainty (2.47%) compared to UV spectroscopy (4.29%), indicating that HPLC provides results with higher confidence and reliability [8].
- Analytical Simplicity vs. Specificity: UV spectroscopy is a rapid, cost-effective technique suitable for simple matrices. However, it lacks the high specificity of HPLC, which can physically separate the analyte from interferents [8] [58].
Experimental Protocols for Method Comparison
To ensure the reliability of the data presented in the comparison, both methods were subjected to rigorous validation following international guidelines. The following workflows and protocols detail the key experiments.
Sample Preparation Workflows
The sample preparation and analysis procedures for UV and HPLC methods differ significantly, impacting time, cost, and complexity.
Detailed Experimental Protocols
Protocol 1: HPLC-UV Method for Piperine Quantification [8]
- Chromatographic Conditions:
- Column: Reverse-phase C18 column.
- Mobile Phase: Methanol and citric acid in water, isocratic or gradient elution.
- Flow Rate: 1.0 mL/min.
- Detection: UV detection at a wavelength specific to piperine (e.g., 343 nm).
- Injection Volume: 10-20 µL.
- Sample Preparation:
- Grind black pepper samples and sieve through a 60-mesh.
- Accurately weigh a portion of the ground pepper.
- Extract with a suitable solvent (e.g., methanol) using sonication or shaking.
- Centrifuge the extract and filter the supernatant through a 0.45 µm membrane filter.
- Inject the filtered solution into the HPLC system.
- Calibration: Prepare a series of piperine standard solutions within the linear range (e.g., 0.8–200 mg/kg) to establish a calibration curve.
Protocol 2: UV Spectroscopy Method for Piperine Quantification [8]
- Instrument Conditions:
- Prepare a blank solution using the extraction solvent.
- Scan standard and sample solutions over a UV range (e.g., 200-400 nm) to identify the wavelength of maximum absorption (λmax) for piperine.
- Sample Preparation:
- Follow steps 1-3 of the HPLC sample preparation.
- After centrifugation and before filtration, dilute the extract to an appropriate concentration within the linear range of the spectrophotometer.
- Measure the absorbance of the sample solution at the predetermined λmax.
- Calibration: Prepare piperine standard solutions and measure their absorbance to create a calibration curve, ensuring the sample absorbance falls within the linear range.
Advanced Optimization and Interference Management
Overcoming Interferences in UV Spectroscopy
UV spectroscopic analysis can be hampered by physical and chemical interferences, but several strategies can mitigate these issues [58]:
- Physical Interferences (Light Scattering): Caused by suspended impurities. Remedied by:
- Filtration or Centrifugation: To clarify the solution.
- Reducing Pathlength: Decreasing the distance between the sample and detector can reduce scattering effects.
- Chemical Interferences (Other Absorbing Compounds): Addressed by:
- Isoabsorbance Measurements: Useful for a single interferent with known absorbance. The absorbance at a wavelength where the interferent absorbs is subtracted from the absorbance at the analytical wavelength.
- Derivative Spectroscopy: A powerful technique that helps resolve overlapping absorption peaks and corrects for baseline shifts by using first or second derivatives of the absorbance spectrum.
- Three-Point Correction: Estimates and subtracts background interference by measuring absorbance at the analytical wavelength and two nearby wavelengths on either side.
The Role of AI and Modern Optimization Tools
The field of analytical method development is being transformed by data science and artificial intelligence (AI). While traditional techniques like Design of Experiments (DoE) are still valuable for systematic optimization [59], new tools are emerging [60] [61]:
- Machine Learning (ML) and Surrogate Optimization: These data-driven approaches can streamline the optimization of complex methods with many variables, reducing the number of experimental steps required.
- AI-Driven HPLC Systems: Hybrid systems that use AI and "digital twins" can autonomously optimize HPLC methods. They predict retention based on molecular structure and then use minimal experiments to calibrate and optimize method variables like flow rate and gradient conditions.
- QSERR Models: Quantitative Structure Enantioselective Retention Relationship (QSERR) models use molecular descriptors to predict the behavior of chiral compounds on specific columns, aiding in the rational design of separation methods.
These advanced techniques represent a shift from empirical, trial-and-error approaches to predictive, in-silico assisted method development.
The Scientist's Toolkit
Table 2: Essential Reagents and Materials for UV and HPLC Analysis
| Item | Function / Application | Example from Research |
|---|---|---|
| HPLC-Grade Solvents | Mobile phase preparation; ensures minimal UV-absorbing impurities and consistent chromatography. | Methanol, Acetonitrile, Water [8] [57] |
| Analytical Standards | Used for calibration and method validation; provides known concentration for quantification. | Piperine standard [8]; Xylitol standard [57] |
| Syringe Filters | Clarification of samples prior to injection; prevents column blockage and system damage. | 0.45 µm HVLP filters [8] |
| Derivatization Reagents | Chemically modifies non-UV-absorbing analytes to introduce a chromophore for detection. | p-Nitrobenzoyl Chloride (PNBC) for xylitol analysis [57] |
| Chromatography Columns | Stationary phase for separating analyte mixtures. | Reverse-phase C18 column [8] [57] |
| Buffer Salts | Modifies mobile phase to control pH and improve separation. | Citric acid [8]; o-Phosphoric acid [59] |
The choice between UV spectroscopy and HPLC is not a matter of one being universally superior, but of selecting the right tool for the specific analytical problem. UV spectroscopy offers a rapid, cost-effective, and simple solution for routine analysis of specific compounds in relatively simple matrices. However, HPLC-UV provides significantly higher specificity, sensitivity, and lower measurement uncertainty, making it the definitive choice for complex samples, trace analysis, and situations demanding high data confidence. The ongoing integration of AI and machine learning is poised to further enhance the efficiency and predictive power of HPLC method development, solidifying its role as an indispensable technique in modern scientific research and drug development.
High-Performance Liquid Chromatography (HPLC) is an indispensable analytical technique in pharmaceutical analysis, providing the precision, sensitivity, and specificity required for drug development, quality control, and regulatory compliance. The technique separates, identifies, and quantifies components in complex mixtures, making it vital for tasks such as purity testing, content uniformity, stability studies, and impurity profiling [62] [63]. The development of a robust HPLC method is a systematic process that hinges on the careful optimization of three fundamental parameters: column chemistry, mobile phase composition, and detection settings. Proper optimization of these parameters enables researchers to achieve the necessary resolution, sensitivity, and accuracy for their specific analytical needs, whether for simple assays or complex formulations containing multiple analytes and potential interferents [64].
This guide objectively compares HPLC with Ultraviolet (UV) spectroscopy, another common analytical technique. The context for this comparison is a broader research thesis on measurement uncertainty, a critical metric for evaluating analytical method performance. Measurement uncertainty defines the confidence in measurement results and is a key differentiator between analytical techniques [8]. We will summarize experimental data and provide detailed methodologies to help researchers and scientists select the most appropriate technique based on their project's requirements for accuracy, precision, and reliability.
Critical Components of HPLC Method Development
Column Chemistry and Selection
The choice of column is the first and one of the most critical decisions in HPLC method development. The column's stationary phase dictates the interaction with analytes and is the primary factor governing separation. The most prevalent type is Reverse Phase HPLC (RP-HPLC), which utilizes a non-polar stationary phase (typically C18-bonded silica) and a polar mobile phase (e.g., water and acetonitrile or methanol). RP-HPLC is ideal for separating non-polar or hydrophobic compounds, including most pharmaceuticals, lipids, and metabolites [65].
Other specialized column chemistries include:
- Normal Phase HPLC (NP-HPLC): Employs a polar stationary phase and a non-polar mobile phase. It is effective for separating polar compounds like carbohydrates, amino acids, and vitamins [65].
- Ion-Exchange Chromatography (IEC): Separates compounds based on ionic interactions. It is widely used for charged molecules like proteins, nucleic acids, and inorganic ions [65].
- Size Exclusion Chromatography (SEC): Separates molecules based on their size and molecular weight. It is crucial for protein analysis and polymer characterization [65].
- Chiral Chromatography: Uses a chiral stationary phase to separate enantiomers, which is essential in pharmaceutical development since different enantiomers can exhibit distinct pharmacological activities [65].
For initial method development, a short (10-15 cm) C18 column with 3 or 5 µm packing particles is recommended for its versatility and shorter analysis time [64].
Mobile Phase Optimization
The mobile phase serves as the solvent that carries the sample through the column. Its composition is a powerful tool for optimizing selectivity and retention. Key considerations include:
- Solvent Strength: In RP-HPLC, the solvent strength is controlled by the concentration of the organic modifier (e.g., acetonitrile or methanol). The goal is to find a concentration that elutes all analytes with capacity factors (k) between 0.5 and 15, balancing resolution and analysis time [64].
- pH and Additives: For analytes that are weak acids or bases, mobile phase pH is a critical parameter. Using buffers (e.g., phosphate or acetate) to control pH can significantly alter retention and peak shape. Ion-pairing reagents may be added for strong acids or bases [64].
- Gradient vs. Isocratic Elution: Isocratic elution uses a constant mobile phase composition and is suitable for simple mixtures. Gradient elution, where the composition changes over time, is necessary for complex samples with a wide range of analyte polarities, as it provides better resolution for later-eluting peaks and shorter run times [64].
A systematic approach involves starting with a binary system, such as acetonitrile/water or methanol/water, and adjusting the strength and pH to achieve the desired separation [64].
Detection Parameters
The detector identifies and quantifies the analytes as they elute from the column. The most common detector is the Ultraviolet (UV) detector. When using UV detection, the wavelength is a key parameter. For maximum sensitivity, the wavelength should be set at the maximum absorbance (λmax) of the target analyte. Wavelengths below 200 nm should generally be avoided due to increased noise and potential solvent interference [64].
For applications requiring higher sensitivity or selectivity, other detectors are available:
- Diode-Array Detection (DAD): Captures full UV-Vis spectra for each peak, enabling peak purity assessment and analyte identification [63].
- Fluorescence Detection: Offers very high sensitivity and selectivity for compounds that are naturally fluorescent or can be derivatized to become so [64].
- Mass Spectrometry (MS): Provides superior sensitivity and enables structural elucidation, making it ideal for trace analysis and impurity identification [62] [63].
The following diagram illustrates the logical workflow for developing and optimizing an HPLC method, integrating the decisions regarding column chemistry, mobile phase, and detection.
Comparative Experimental Data: HPLC vs. UV Spectroscopy
To objectively compare performance, we examine experimental data from studies that directly applied both UV spectroscopy and HPLC to the same analytical problems. The following table summarizes key validation parameters from two such studies, one analyzing piperine in black pepper and the other analyzing favipiravir in pharmaceutical formulations [8] [29].
Table 1: Comparative Performance Data from Validation Studies
| Analytical Parameter | Piperine in Black Pepper [8] | Favipiravir in Formulations [29] | ||
|---|---|---|---|---|
| Technique | UV Spectroscopy | HPLC-UV | UV Spectroscopy | HPLC-UV |
| Linearity (R²) | Good | Good | >0.998 (implied) | >0.998 (implied) |
| Accuracy (% Recovery) | 96.7 - 101.5% | 98.2 - 100.6% | 90 - 110% (accepted) | 90 - 110% (accepted) |
| Precision (% RSD) | 0.59 - 2.12% | 0.83 - 1.58% | Not Specified | ≤ 2% (accepted) |
| Limit of Detection (LOD) | 0.65 (unitless) | 0.23 (unitless) | Calculated via slope/error | Calculated via slope/error |
| Limit of Quantification (LOQ) | Not Specified | Not Specified | Calculated via slope/error | Calculated via slope/error |
| Measurement Uncertainty | 4.29% (at 49.481 g/kg) | 2.47% (at 34.819 g/kg) | Not Specified | Not Specified |
A broader comparison of the general characteristics, advantages, and limitations of each technique is provided in the table below.
Table 2: General Comparison of UV Spectroscopy and HPLC
| Aspect | UV Spectroscopy | HPLC |
|---|---|---|
| Cost & Equipment | Low cost; simple setup [63] | High cost; complex instrumentation [63] |
| Selectivity/Specificity | Limited; prone to spectral overlaps from excipients or impurities [63] | High; excellent separation of components, even in complex mixtures [63] [29] |
| Sensitivity | Good for simple assays with strong chromophores [63] | Superior; capable of detecting and quantifying low-level impurities [63] |
| Sample Preparation | Minimal [63] | Often more complex; may require extraction, filtration, etc. [63] [64] |
| Analysis Speed | Fast [63] | Moderate; method length varies [63] |
| Best Use Cases | Routine QC of simple, single-component samples [63] | Complex formulations, impurity profiling, stability-indicating methods [63] |
| Key Limitation | Requires a chromophore; cannot analyze mixtures without separation [63] | Costly, requires skilled operation, high solvent consumption [63] |
Detailed Experimental Protocols
Protocol 1: Determination of Piperine by HPLC-UV and UV Spectroscopy
This protocol is derived from a study comparing the measurement uncertainty of both methods for quantifying piperine in black pepper [8].
A. Sample Preparation:
- Collect black pepper samples and grind them using a blender.
- Sieve the ground powder through a 60-mesh screen.
- For analysis, prepare standard and sample solutions in a suitable solvent (e.g., methanol).
B. UV Spectroscopy Method:
- Instrumentation: Standard UV-Vis spectrophotometer.
- Analysis: Scan the standard and sample solutions to determine the maximum absorption wavelength (λmax) for piperine. Measure the absorbance of sample solutions at this λmax.
- Quantification: Construct a calibration curve using piperine standard solutions and use it to determine the concentration in unknown samples.
C. HPLC-UV Method:
- Instrumentation: HPLC system equipped with a UV detector and a C18 column.
- Mobile Phase: Use a mixture of water and acetonitrile. The exact ratio should be optimized for separation (e.g., a study used sodium acetate buffer and acetonitrile 85:15, v/v) [29].
- Chromatographic Conditions:
- Injection & Elution: Inject the standard and sample solutions. The run time is typically 10 minutes or as needed to elute the target compound.
D. Method Validation:
- Validate both methods by assessing specificity, linearity, LOD, LOQ, accuracy, and precision according to ICH guidelines [8] [66].
- Calculate measurement uncertainty by combining all uncertainty sources from the sample preparation, standard preparation, and instrument measurement [8].
Protocol 2: Fast HPLC for Dissolution Testing
This protocol outlines the development of an ultrafast HPLC method for dissolution testing of pharmaceutical products, where speed is critical [67].
A. Define Goal and Constraints:
- Target a very short analysis time (e.g., a column dead time, t₀, of ~4 seconds for a total run time of about 30 seconds).
- Determine the maximum operating pressure (P_max) of the HPLC system.
B. Performance Optimization:
- Select Particle Size: For ultrafast separations, use columns packed with small particles (e.g., 1.8 µm to 1.0 µm).
- Optimize Column Length and Flow Rate: Use a short column (e.g., 30-50 mm). Calculate the optimal linear velocity and column length using a two-parameter optimization approach to achieve the highest plate count within the target time and pressure constraints [67].
- Adjust Temperature: Operate at a higher temperature (e.g., 40-60°C) to reduce mobile phase viscosity, which allows for higher flow rates at lower pressures.
C. Method Implementation:
- Column: Select a narrow-bore column (e.g., 2.1 mm diameter) packed with sub-2µm particles.
- Mobile Phase: Use an isocratic mixture appropriate for the API (e.g., a buffer/acetonitrile mix).
- Conditions: Set the flow rate and temperature to the optimized values. The method should elute the API within the target time while maintaining adequate resolution from any excipient peaks.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential Materials for HPLC Method Development and Analysis
| Item | Function / Purpose | Examples / Notes |
|---|---|---|
| HPLC System | Separates, identifies, and quantifies components in a sample. | Consists of pump, autosampler, column oven, and detector [64]. |
| C18 Column | The most common reverse-phase stationary phase for separating non-polar to moderately polar compounds. | Inertsil ODS-3 [29]; Symmetry C18 [66]. Length and particle size vary by application. |
| Acetonitrile & Methanol | HPLC-grade organic solvents used as the mobile phase or its component. | Ensure high purity to reduce baseline noise and UV background [29] [64]. |
| Water (HPLC Grade) | The polar component of the mobile phase in reverse-phase HPLC. | Must be ultra-pure, typically from a Milli-Q water purification system [8] [66]. |
| Buffer Salts | Used to prepare mobile phase buffers for controlling pH and ionic strength. | Sodium acetate [29], orthophosphoric acid for pH adjustment [66]. |
| Standard Reference | A high-purity compound used for calibration and identification. | Piperine standard [8], Diclofenac Sodium [66], Favipiravir [29]. |
| Syringe Filters | For clarifying and purifying sample solutions before injection into the HPLC system. | 0.45 µm or 0.22 µm pore size, made from nylon or PVDF [8] [29]. |
HPLC and UV spectroscopy are both powerful techniques for pharmaceutical analysis, but they serve different purposes. UV spectroscopy is a rapid, simple, and cost-effective solution for routine quality control of simple, single-component samples where the analyte possesses a strong chromophore. However, its main drawbacks are lower specificity and higher measurement uncertainty when analyzing complex mixtures.
HPLC, particularly HPLC-UV, offers superior specificity, sensitivity, and lower measurement uncertainty, making it the definitive choice for complex formulations, impurity profiling, and stability-indicating assays. The key trade-offs are higher instrument cost, operational complexity, and longer analysis time. The experimental data clearly demonstrates that HPLC provides more accurate and reliable results where separation from interferents is crucial. For researchers and drug development professionals, the choice between these techniques should be guided by the specific analytical requirements, balancing the need for speed and cost against the imperative for specificity and data certainty.
The quantitative analysis of chemical substances is a cornerstone of pharmaceutical development and forensic science. For decades, high-performance liquid chromatography (HPLC) has been the undisputed gold standard for quantification, prized for its high precision and sensitivity [30] [68]. However, HPLC relies on costly reference standards, toxic solvents, and specific methods for each analyte [30]. In parallel, UV spectrophotometry offers a simple and rapid alternative but suffers from low specificity, making it unsuitable for analyzing complex mixtures without prior separation [68] [8]. Within this landscape, Nuclear Magnetic Resonance (NMR) spectroscopy is recognized as a powerful Category A identification technique [30] that is inherently quantitative, as the signal intensity is directly proportional to the number of nuclei generating it [69]. Historically, its use in routine quantification was limited by the high cost and large size of conventional superconducting magnet systems [30].
The advent of benchtop NMR spectrometers has begun to change this paradigm. These compact, cost-effective instruments are far more accessible but operate at lower magnetic fields, which leads to reduced spectral resolution and increased peak overlap compared to their high-field counterparts [30] [70]. This complexity makes traditional analysis of benchtop NMR data challenging. To unlock its quantitative potential, advanced data processing techniques are required. Among these, Quantum Mechanical Modeling (QMM), also known as quantum mechanics-total-line-shape fitting, has emerged as a powerful solution. This guide objectively compares the performance of benchtop NMR enhanced with QMM against traditional HPLC and UV methods, providing researchers with the data needed to evaluate its role as a complementary analytical technique.
Core Technologies and Comparative Fundamentals
To understand the value of benchtop NMR with QMM, it is essential to grasp the fundamental principles and limitations of the established techniques.
UV Spectrophotometry operates on the Beer-Lambert law, measuring the absorption of ultraviolet light by a sample at a specific wavelength [68]. It is best suited for clear, single-component solutions, as excipients or impurities that absorb at the same wavelength can cause significant interference and inaccurate results [68]. Its primary advantages are speed, simplicity, and low cost [68].
HPLC-UV separates the components of a mixture based on their differential distribution between a stationary and a mobile phase before quantifying them via UV detection [68]. This separation grants it high specificity for multi-analyte determination, impurity profiling, and stability studies, making it the pharmacopeial standard for these applications [68] [8]. Its main drawbacks are longer analysis times, higher operational costs, and a dependence on specific calibration standards for each compound [30].
Benchtop NMR with QMM leverages the intrinsic quantitative nature of NMR signal. QMM software, such as Q2NMR, tackles the issue of spectral overlap in low-field spectra by using known NMR parameters (e.g., chemical shifts, coupling constants) to generate a complete quantum-mechanical model of the spectrum [30]. This ideal spectrum is then fitted to the experimental data, effectively deconvoluting overlapping peaks and allowing for the simultaneous identification and quantification of multiple compounds, including the active ingredient, adulterants, and impurities, in a single, rapid measurement [30].
The table below summarizes the key differences between these core technologies.
Table 1: Fundamental Comparison of UV, HPLC, and Benchtop NMR Techniques
| Feature | UV Spectrophotometry | HPLC-UV | Benchtop NMR with QMM |
|---|---|---|---|
| Principle | UV light absorption [68] | Chromatographic separation + UV detection [68] | Nuclear spin resonance with quantum mechanical spectral fitting [30] |
| Specificity | Low | High | Highest (Category A technique) [30] |
| Sample Type | Single-component, clear solutions [68] | Complex mixtures | Complex mixtures (solid & liquid) |
| Quantitative Nature | Requires calibration | Requires analyte-specific calibration [30] | Inherently quantitative; can use a single universal standard [30] [69] |
| Key Limitation | Interference from other chromophores [68] | Cost, solvent use, need for multiple standards [30] | Lower sensitivity and resolution vs. high-field NMR [30] |
Performance Comparison: Quantitative Data and Validation
Direct, head-to-head studies demonstrate that benchtop NMR with QMM can achieve a level of accuracy and precision that is competitive with HPLC for the analysis of complex mixtures.
A pivotal 2025 study directly compared benchtop NMR and HPLC-UV for quantifying methamphetamine hydrochloride in binary and ternary mixtures containing cutting agents. The root mean square error (RMSE) for benchtop NMR using the QMM method was as low as 1.3 mg analyte per 100 mg of sample. When the same set of samples was analyzed, the RMSE for QMM was 2.1, compared to 1.1 for HPLC-UV, indicating comparable and highly acceptable accuracy for the NMR method [30]. Another study on carbohydrate analysis in a traditional Chinese medicine injection found that qNMR (both internal and external standard methods) and multiple HPLC methods (PMP-HPLC, RID, ELSD) showed no significant statistical differences in the quantified levels of fructose, glucose, sucrose, and maltose [69]. Both techniques demonstrated excellent correlation coefficients (>0.999), precision (RSD generally <2%), and accuracy [69].
The performance of a method must also be judged by its validation parameters. The following table compiles key validation data from various studies, illustrating the reliability of a well-configured benchtop NMR system.
Table 2: Comparison of Method Validation Parameters Across Techniques
| Method & Application | Accuracy (%) | Precision (RSD%) | Linearity (R²) | Key Reference |
|---|---|---|---|---|
| Benchtop qNMR (Purity) | 98 - 102 | < 0.6 % | > 0.9999 | [71] |
| HPLC-UV (Favipiravir) | 99.6 - 100.1 | < 1.6 % | > 0.999 | [72] |
| UV Spectrophotometry (Favipiravir) | 99.8 - 100.5 | < 2.1 % | > 0.999 | [72] |
| HPLC-UV (Piperine) | 98.2 - 100.6 | 0.8 - 1.6 % | > 0.999 | [8] |
| UV Spectrophotometry (Piperine) | 96.7 - 101.5 | 0.6 - 2.1 % | > 0.999 | [8] |
Furthermore, a comparison of measurement uncertainty for piperine analysis found that HPLC-UV had a lower expanded uncertainty (2.47%) compared to UV spectroscopy (4.29%), confirming HPLC as a more precise and reliable technique [8]. While a direct uncertainty comparison for benchtop NMR was not provided in the search results, its demonstrated accuracy and precision in the aforementioned studies suggest it can deliver highly certain results, positioning it as a robust complementary technique.
Experimental Protocols: Implementing Benchtop NMR with QMM
For researchers seeking to implement this technology, understanding the standard experimental workflow is crucial. The following protocol is adapted from the 2025 study on methamphetamine analysis and other relevant sources [30] [71].
Detailed Methodology
Sample Preparation:
- Weigh the analyte and any cutting agents or impurities accurately to prepare binary and ternary mixtures across the desired purity range (e.g., 10-90 mg per 100 mg of sample) [30].
- For quantitative purity analysis, use an internal standard (e.g., maleic acid) with a known purity. Precisely weigh the sample and the internal standard [71].
- Dissolve the mixture in a deuterated solvent (e.g., D₂O) at a typical concentration range of 15-75 mg/mL to ensure a strong signal-to-noise ratio [71]. Filter the solution if necessary.
NMR Data Acquisition:
- Use a 60-MHz or similar benchtop NMR spectrometer.
- Transfer the sample to a standard NMR tube and insert it into the spectrometer.
- Acquire the ¹H NMR spectrum with sufficient scans to achieve a good signal-to-noise ratio. Key acquisition parameters include a 90° pulse angle and a relaxation delay (d1) of at least 5 times the longitudinal relaxation time (T₁) of the nuclei being quantified to ensure complete relaxation and quantitative accuracy [30] [69].
Spectral Processing with QMM:
- Transfer the free induction decay (FID) data to software capable of QMM analysis (e.g., Q2NMR).
- Input the molecular structures of all compounds suspected to be in the mixture.
- The QMM algorithm will use quantum mechanics to calculate the theoretical spectrum based on parameters like chemical shifts and J-couplings.
- The software then performs a least-squares fitting of this theoretical model to the entire experimental spectrum. This fit directly provides the mole fractions and subsequently the concentrations or purities of all components simultaneously [30].
Figure 1: The QMM-Enhanced Benchtop NMR Workflow. This diagram outlines the key steps from sample preparation to final result.
The Scientist's Toolkit: Essential Reagents and Materials
Successful implementation of benchtop NMR quantification, particularly with the QMM method, relies on a set of key reagents and materials.
Table 3: Essential Research Reagent Solutions for Benchtop NMR with QMM
| Item | Function & Importance | Examples / Specifications |
|---|---|---|
| Benchtop NMR Spectrometer | The core instrument for data acquisition; permanent magnet design allows for compact size and easy operation. | 60-MHz spectrometer (e.g., Spinsolve) [30] [71] |
| QMM Software | Advanced data processing platform that deconvolutes overlapping peaks in low-field spectra for accurate quantification. | Q2NMR software [30] |
| Deuterated Solvent (e.g., D₂O) | Provides a signal for the NMR spectrometer to lock onto, ensuring spectral stability, and dissolves the sample. | D₂O, Methanol-d4 [71] [69] |
| Quantitative NMR Standard | A substance of known purity and proton count used as an internal or external reference for concentration calculation. | Maleic Acid, Methylsulfonylmethane (MSM) [71] |
| Analytical Balance | Critical for the precise weighing of samples and internal standards, a major source of uncertainty in qNMR. | High-precision balance (e.g., MettlerToledo) [72] |
| Certified Reference Materials | High-purity analytes, adulterants, and impurities used to build and validate the QMM spectral library. | Methamphetamine HCl, Caffeine, Pseudoephedrine HCl [30] |
Discussion: Positioning QMM-NMR in the Analytical Workflow
The experimental data shows that while HPLC-UV maintains a slight edge in absolute precision, benchtop NMR with QMM offers a powerful set of advantages that make it an excellent complementary technique [30]. Its key strength lies in its ability to simultaneously identify and quantify multiple components in a mixture without requiring analyte-specific calibration curves [30]. This "universal calibration" drastically reduces the need for expensive and sometimes unavailable certified reference standards for every compound, a significant benefit for analyzing novel psychoactive substances or complex natural products like carbohydrates in traditional medicines [30] [69].
Furthermore, benchtop NMR is more robust and environmentally friendly than HPLC, as it uses minimal amounts of deuterated solvents compared to the continuous flow of toxic solvents required for HPLC [30]. The combination of benchtop NMR with chemometric tools like Partial Least Squares (PLS) regression has also proven highly effective for determining parameters like molecular weight in lignin, a task that would otherwise require relative methods like GPC [70]. This demonstrates the platform's versatility beyond simple purity assays.
The following diagram illustrates the strategic position of benchtop NMR with QMM relative to other techniques based on specificity and quantitative power.
Figure 2: Analytical Technique Positioning. This plot positions common techniques based on their general specificity and quantitative power, highlighting the unique space occupied by advanced benchtop NMR.
The integration of Quantum Mechanical Modeling with benchtop NMR spectroscopy represents a significant advancement in analytical chemistry. It successfully addresses the primary limitation of low-field NMR—spectral overlap—unlocking its full potential for both identification and quantification. While HPLC-UV remains the more precise technique for standardized, high-throughput quantification of specific target analytes, benchtop NMR with QMM offers a complementary, robust, and information-rich alternative. It is particularly powerful for the simultaneous analysis of multi-component mixtures, forensic drug profiling, and applications where solvent use and standard availability are limiting factors. As these instruments and software algorithms continue to evolve, benchtop NMR is poised to become an indispensable tool in research and quality control laboratories, providing a unique balance of performance, accessibility, and analytical insight.
The pharmaceutical industry is undergoing a significant paradigm shift, moving away from traditional, compliance-driven quality control methods toward a more systematic, science-based approach known as Quality by Design (QbD). When applied to analytical method development, this approach is termed Analytical Quality by Design (AQbD). AQbD emphasizes deep product and process understanding based on sound science and quality risk management, rather than relying solely on end-product testing [73] [74]. This systematic approach begins with predefined objectives and emphasizes understanding and control, based on robust scientific principles [73].
The International Conference on Harmonisation (ICH) guidelines Q8 (R2), Q9, Q10, Q11, and the newer Q14 provide the framework for implementing QbD and AQbD principles [74]. The fundamental goal of AQbD is to enhance method robustness and ensure regulatory flexibility. It allows analytical methods to operate within a defined Method Operable Design Region (MODR), which is a multidimensional combination of analytical factors that have been demonstrated to provide suitable method performance [73] [74]. By developing methods with built-in robustness, pharmaceutical companies can reduce out-of-trend (OOT) and out-of-specification (OOS) results, thereby improving product quality and regulatory outcomes [73].
The AQbD Framework: A Systematic Methodology
Core Principles and Workflow
Implementing AQbD involves a structured, systematic workflow that parallels the product QbD process. The table below outlines the key stages in the AQbD paradigm [73]:
Table 1: Stages of Implementation for Product QbD and Analytical QbD
| Stage | Product QbD | Analytical QbD |
|---|---|---|
| Stage 1 | Define Quality Target Product Profile (QTPP) | Define Analytical Target Profile (ATP) |
| Stage 2 | Identify Critical Quality Attributes (CQAs) | Identify Critical Method Attributes |
| Stage 3 | Perform Risk Assessment | Perform Risk Assessment |
| Stage 4 | Establish Design Space | Establish Method Operable Design Region (MODR) |
| Stage 5 | Implement Control Strategy | Implement Control Strategy |
| Stage 6 | Manage Life Cycle | Manage Life Cycle |
The process begins with defining the Analytical Target Profile (ATP), which is a prospective summary of the required quality characteristics of an analytical method. The ATP defines the purpose of the method and its performance requirements [73]. Following this, critical method attributes are identified, and a risk assessment is conducted to determine which method parameters significantly impact the method's performance. This risk assessment guides subsequent optimization experiments [74].
AQbD vs. Traditional Workflow
The following diagram illustrates the fundamental difference between the traditional OFAT approach and the systematic AQbD workflow.
The core difference lies in the optimization strategy. The traditional One-Factor-at-a-Time (OFAT) approach optimizes one parameter while holding others constant, which often yields a narrow robust region and fails to capture parameter interactions [73] [74]. In contrast, AQbD employs multivariate Design of Experiments (DoE) to scientifically explore the interaction of all critical parameters simultaneously, leading to the establishment of a robust MODR [73]. This MODR provides regulatory flexibility, as movement within this defined space is not considered a change and does not require revalidation [73] [74].
Comparative Analysis: UV Spectroscopy vs. HPLC-UV
To objectively evaluate the performance of different analytical techniques, a direct comparison of validation parameters and measurement uncertainty is essential. The following analysis uses experimental data from studies that implemented rigorous, ICH-compliant validation protocols.
Performance Comparison for Piperine Analysis
A comprehensive study directly compared UV spectroscopy and HPLC-UV for the determination of piperine in black pepper, estimating performance parameters and measurement uncertainties to identify the more efficient method [8].
Table 2: Validation Parameters for Piperine Quantification: UV vs. HPLC-UV [8]
| Validation Parameter | UV Spectroscopy | HPLC-UV |
|---|---|---|
| Specificity | Good | Good |
| Linearity (R²) | Good | Good |
| LOD | 0.65 µg/mL | 0.23 µg/mL |
| LOQ | Information Not Specified | Information Not Specified |
| Accuracy (%) | 96.7 – 101.5 | 98.2 – 100.6 |
| Precision (% RSD) | 0.59 – 2.12 | 0.83 – 1.58 |
| Measurement Uncertainty (k=2) | 4.29% (at 49.481 g/kg) | 2.47% (at 34.819 g/kg) |
Key Findings:
- Sensitivity: HPLC-UV demonstrated superior sensitivity, with a lower Limit of Detection (LOD) (0.23 µg/mL) compared to UV spectroscopy (0.65 µg/mL) [8].
- Accuracy and Precision: Both methods showed excellent accuracy (close to 100%) and acceptable precision (RSD < 2%). The precision for UV was slightly better in the reported study, though this is highly method-dependent [8].
- Measurement Uncertainty: A critical differentiator was measurement uncertainty. The HPLC-UV method exhibited significantly lower uncertainty (2.47%) compared to UV spectroscopy (4.29%), making it a more reliable and accurate technique for quantitative analysis [8].
Performance Comparison for Repaglinide Analysis
Another study developing methods for the antidiabetic drug repaglinide reinforces these findings, demonstrating that both techniques can be optimized for routine quality control [1].
Table 3: Validation Parameters for Repaglinide Quantification [1]
| Validation Parameter | UV Spectroscopy | HPLC-UV |
|---|---|---|
| Linearity Range | 5 – 30 µg/mL | 5 – 50 µg/mL |
| Linearity (R²) | > 0.999 | > 0.999 |
| Precision (% RSD) | < 1.50 | Better than UV method |
| Accuracy (% Recovery) | 99.63 – 100.45 | 99.71 – 100.25 |
Key Findings:
- Linearity and Accuracy: Both methods demonstrated excellent linearity and accuracy, with recovery rates very close to 100% [1].
- Precision: The HPLC method was found to be "highly precise" compared to the UV method, which simply met the acceptance criteria [1].
- Working Range: The HPLC method offered a wider linear range, making it suitable for a broader array of sample concentrations [1].
Detailed Experimental Protocols
To ensure reproducibility and provide a clear basis for comparison, the detailed experimental protocols from the cited studies are outlined below.
- Instrumentation: High-performance liquid chromatography system coupled with an ultraviolet detector.
- Column: C18 reversed-phase column (a common choice in AQbD implementations [75]).
- Detection Wavelength: 360 nm.
- Mobile Phase: The specific composition was optimized; AQbD principles would involve defining an MODR for the mobile phase composition, pH, and buffer concentration.
- Sample Preparation: Black pepper samples were ground, sieved, and extracted. The solution was filtered before injection.
- Validation: The method was validated for specificity, LOD, LOQ, linearity, accuracy, and precision according to ICH guidelines. Measurement uncertainty was estimated according to the Eurachem Guide and NIST guidelines.
- Instrumentation: Ultraviolet-Visible spectrophotometer.
- Solvent: Methanol.
- Detection Wavelength: The wavelength of maximum absorption for piperine was used.
- Sample Preparation: Black pepper samples were prepared via extraction to dissolve the piperine.
- Validation: The method was validated using the same parameters as the HPLC method (specificity, LOD, LOQ, etc.), allowing for a direct comparison of performance.
This protocol for simultaneous determination of neuromodulating drugs (Piracetam, Gabapentin, Levetiracetam) exemplifies a robust AQbD-developed method.
- Instrumentation: HPLC-UV system (Shimadzu).
- Column: Inertsil ODS-3 C18 (250 mm × 4.6 mm, 5 µm).
- Mobile Phase: Isocratic elution with Methanol:Water (15:85, v/v).
- Flow Rate: 1.5 mL/min.
- Detection: 210 nm.
- Temperature: Ambient.
- Validation: The method was validated per ICH guidelines over ranges of 30.0–1000.0 µg/mL for GBP and 10.0–100.0 µg/mL for LEV and PIR.
The Scientist's Toolkit: Essential Reagents and Materials
The following table lists key materials and reagents commonly used in developing and executing UV and HPLC-UV methods, based on the experimental protocols provided.
Table 4: Essential Research Reagents and Materials for Analytical Method Development
| Item | Function / Application | Example from Protocols |
|---|---|---|
| HPLC-Grade Methanol & Acetonitrile | Organic solvent for mobile phase preparation and sample extraction. | Used in mobile phase for repaglinide [1] and piperine analysis [8]. |
| HPLC-Grade Water | Aqueous component of mobile phase; sample diluent. | Used in all cited HPLC protocols [1] [8] [76]. |
| Analytical Reference Standards | Certified pure compounds for calibration and method validation. | Piperine standard [8]; Repaglinide standard [1]. |
| Buffering Salts & pH Adjusters | Control pH of mobile phase to improve separation and peak shape. | Orthophosphoric acid to adjust pH to 3.5 [1]; Citric acid for mobile phase [8]. |
| Syringe Filters (0.45 µm or 0.22 µm) | Clarify sample solutions by removing particulate matter before injection into the HPLC system. | 0.45 µm HVLP filters [8]; 0.45 µm membrane filter [76]. |
| C18 Reversed-Phase Chromatography Column | The stationary phase for separation; the most common column type in pharmaceutical analysis. | Agilent TC-C18 column [1]; Inertsil ODS-3 C18 column [76]. |
Understanding and controlling sources of uncertainty is fundamental to building robust analytical methods.
Uncertainty in HPLC-UV
In HPLC-UV, uncertainty propagates through several steps [5]:
- Injection Step: Uncertainty in the injected sample volume is a major contributor.
- Separation Step: Uncertainties related to retention volume, number of theoretical plates, temperature, and flow rate.
- Detection Step: Uncertainty in the measurement of absorbance.
Studies have shown that the uncertainty in injection volume contributes the greatest uncertainty to the final measured peak area [5]. Furthermore, the uncertainty of the eluate concentration at the detector is not zero, even if the injected standard concentration is known exactly [5].
The Role of AQbD in Managing Uncertainty
The AQbD framework directly addresses these uncertainties through systematic robustness testing. As per ICH Q2(R2), robustness is "a measure of [the method's] capacity to meet the expected performance requirements during normal use" and is tested by deliberate variations of method parameters [74]. By using DoE to understand the effect of these variations, an MODR is established where the method performs satisfactorily, thereby controlling and minimizing the impact of uncertainties on the final analytical result.
The choice between UV spectroscopy and HPLC-UV must be guided by the Analytical Target Profile. UV spectroscopy offers a rapid, simple, and cost-effective solution for applications where high sensitivity is not critical and sample matrices are simple. However, for methods requiring high sensitivity, specificity in complex matrices, and lower measurement uncertainty, HPLC-UV is the unequivocally superior technique.
The implementation of Analytical Quality by Design principles is pivotal for building robustness into either technique. By moving from the traditional OFAT approach to a systematic, risk-based, and multivariate methodology, scientists can develop methods with a well-understood Method Operable Design Region. This not only reduces the frequency of OOS and OOT results but also provides regulatory flexibility, ultimately ensuring higher product quality and more efficient pharmaceutical development and manufacturing.
Validation Parameters and Decision Framework for Method Selection
Within pharmaceutical development and quality control, analytical methods must be rigorously demonstrated to be suitable for their intended purpose. The International Council for Harmonisation (ICH) guideline Q2(R2) on the validation of analytical procedures provides a foundational framework for this process, outlining key parameters that must be evaluated [77]. Among these, linearity, precision, accuracy, and specificity are critical for ensuring that methods reliably measure the analyte of interest.
This guide objectively compares the performance of two fundamental analytical techniques—Ultraviolet (UV) spectroscopy and High-Performance Liquid Chromatography (HPLC)—against these ICH validation parameters. The comparison is framed within broader research on measurement uncertainty, providing scientists and drug development professionals with the experimental data necessary to make informed method selection decisions.
Core ICH Validation Parameters Explained
The ICH Q2(R2) guideline defines the validation characteristics required for analytical procedure registration applications. The following parameters are central to demonstrating method suitability [77] [78]:
- Specificity: The ability to assess unequivocally the analyte in the presence of components that may be expected to be present, such as impurities, degradation products, or matrix components. It is the cornerstone that ensures a method measures only what it should [78].
- Accuracy: The closeness of agreement between the value which is accepted as a conventional true value or an accepted reference value and the value found. It expresses the systematic error of a measurement, often reported as percent recovery [78].
- Precision: The closeness of agreement between a series of measurements obtained from multiple sampling of the same homogeneous sample under the prescribed conditions. It is typically subdivided into repeatability (intra-assay precision) and intermediate precision (variations between different days, analysts, or equipment) [78] [79].
- Linearity: The ability of the method, within a given range, to obtain test results that are directly proportional to the concentration of the analyte. The Range is the interval between the upper and lower concentrations for which suitable levels of precision, accuracy, and linearity have been demonstrated [78] [79].
Comparative Analysis of UV Spectroscopy and HPLC
Direct comparisons of UV and HPLC methods, applied to the same analyte, reveal distinct performance differences aligned with their fundamental principles. HPLC separates a sample into its individual components, while UV spectroscopy measures the total absorbance of a solution.
Case Study 1: Analysis of Repaglinide
A study developing methods for the antidiabetic drug repaglinide in tablets validated both UV and reversed-phase HPLC (RP-HPLC) methods [1].
Table 1: Validation Data for Repaglinide Analysis [1]
| Validation Parameter | UV Spectrophotometry | RP-HPLC Method |
|---|---|---|
| Linearity Range | 5–30 μg/mL | 5–50 μg/mL |
| Correlation (r²) | > 0.999 | > 0.999 |
| Precision (%RSD) | < 1.50% | Better than UV method |
| Accuracy (% Recovery) | 99.63–100.45% | 99.71–100.25% |
| Specificity | Assessed via spectrum in 200-400 nm | Assessed via chromatographic separation |
Both methods demonstrated excellent linearity and accuracy. However, the HPLC method showed superior precision and a wider linear range. The fundamental difference lies in specificity: while the UV method relied on the absence of interfering bands at 241 nm, the HPLC method confirmed specificity by demonstrating that excipients did not produce interfering peaks at the retention time of repaglinide [1].
Case Study 2: Analysis of Piperine in Black Pepper
A 2022 study directly compared UV and HPLC-UV methods for piperine quantification, including an assessment of measurement uncertainty [8].
Table 2: Validation Data for Piperine Analysis [8]
| Validation Parameter | UV Spectrophotometry | HPLC-UV Method |
|---|---|---|
| Linearity | Good | Good |
| LOD | 0.65 (relative value) | 0.23 (relative value) |
| LOQ | Not specified | Not specified |
| Accuracy (% Recovery) | 96.7–101.5% | 98.2–100.6% |
| Precision (%RSD) | 0.59–2.12% | 0.83–1.58% |
| Measurement Uncertainty | 4.29% (at 49.481 g/kg) | 2.47% (at 34.819 g/kg) |
The study concluded that HPLC was more sensitive (as evidenced by the lower LOD) and more accurate. A key finding was the significantly lower measurement uncertainty for the HPLC method, making it more reliable for obtaining precise and dependable results [8].
Case Study 3: Analysis of Levofloxacin in a Complex Scaffold
Research on levofloxacin released from a mesoporous silica/nano-hydroxyapatite composite scaffold highlighted the impact of sample complexity [18]. While both methods showed excellent linearity (R² > 0.999), their accuracy profiles differed significantly in the presence of the scaffold matrix. The recovery rates for a medium concentration (25 μg/mL) were 110.96% for HPLC versus 99.50% for UV-Vis, suggesting that the HPLC method, with an internal standard and separation step, was better able to compensate for matrix effects [18]. The study identified HPLC as the preferred method for accurate determination in this complex drug-delivery system.
Experimental Protocols for Method Comparison
To ensure reliable comparison data, studies follow standardized experimental protocols and validation as per ICH guidelines.
Detailed Methodology for HPLC Analysis
A typical protocol, as used for repaglinide, includes the following steps [1]:
- Instrumentation: Agilent 1120 Compact LC system with a UV detector.
- Column: Agilent TC-C18 (250 mm × 4.6 mm, 5 μm particle size).
- Mobile Phase: Methanol and water (80:20 v/v, pH adjusted to 3.5 with orthophosphoric acid).
- Flow Rate: 1.0 mL/min.
- Detection Wavelength: 241 nm.
- Sample Preparation: Tablets are powdered, dissolved in methanol, sonicated, filtered, and diluted with the mobile phase.
Detailed Methodology for UV Spectroscopy Analysis
The corresponding UV method for the same analyte was [1]:
- Instrumentation: Shimadzu 1700 Double beam UV-Vis spectrophotometer.
- Solvent: Methanol.
- Detection Wavelength: 241 nm.
- Sample Preparation: Tablets are powdered, dissolved in methanol, sonicated, filtered, and diluted with methanol.
Measurement Uncertainty: A Decisive Factor
Measurement uncertainty is a quantitative parameter that characterizes the dispersion of values that could reasonably be attributed to the measurand. It provides a crucial metric for comparing methods. As demonstrated in the piperine study, the HPLC-UV method exhibited a nearly two-fold lower expanded uncertainty (2.47%) compared to the UV method (4.29%) [8]. This lower uncertainty is directly tied to HPLC's superior specificity and precision, which reduce systematic and random errors, making it the more reliable choice for critical applications.
The Scientist's Toolkit: Essential Research Reagents and Materials
The following table details key materials and reagents commonly used in these analytical methods, based on the cited protocols.
Table 3: Essential Research Reagents and Materials
| Item | Function | Example from Research |
|---|---|---|
| HPLC-Grade Methanol | Mobile phase component; solvent for sample preparation. | Used in mobile phase for repaglinide [1] and quercetin analysis [22]. |
| HPLC-Grade Water | Mobile phase component to adjust eluting strength. | Used in mobile phase for repaglinide HPLC [1]. |
| C18 Chromatography Column | Stationary phase for reverse-phase separation. | Agilent TC-C18 column [1]; Sepax BR-C18 column [18]. |
| Reference Standard | Provides the "true value" for calibration, accuracy, and identification. | Repaglinide from USV Lab [1]; Piperine from Sigma-Aldrich [8]. |
| Orthophosphoric Acid | Mobile phase modifier to control pH for peak symmetry. | Used to adjust mobile phase to pH 3.5 for repaglinide [1]. |
| Ultrasonic Cleaner | To dissolve samples and degas solvents. | Used for dissolving repaglinide tablet powder [1]. |
| Syringe Filters (0.45 μm) | To remove particulate matter from samples before injection. | HVLP filters used in piperine analysis [8]. |
Workflow and Decision Pathway
The following diagram illustrates the logical process for selecting and validating an analytical method based on ICH parameters and sample complexity.
The comparative analysis of UV spectroscopy and HPLC against ICH validation parameters consistently demonstrates a clear performance trade-off. UV spectroscopy offers simplicity, speed, and lower operational cost, making it a viable option for applications involving simple matrices where specificity is not a primary concern.
In contrast, HPLC provides superior specificity, precision, and lower measurement uncertainty, especially in complex matrices. The ability to separate the analyte from impurities and excipients makes HPLC the unequivocally more reliable and robust technique for critical applications in drug development and quality control. The choice between them should be guided by the sample complexity and the required level of analytical confidence, with HPLC being the preferred method for compliance with rigorous ICH standards.
In the field of analytical chemistry, the choice between Ultraviolet (UV) spectroscopy and High-Performance Liquid Chromatography (HPLC) is a fundamental consideration in method development for pharmaceutical analysis. A critical component of this decision is a rigorous statistical comparison of the performance characteristics of the two techniques, grounded in the principles of measurement uncertainty. This guide provides an objective, data-driven comparison of UV and HPLC methods, employing paired t-tests and F-tests to evaluate their agreement across key analytical figures of merit. The analysis synthesizes experimental data from multiple published studies to inform researchers, scientists, and drug development professionals in their selection of an appropriate analytical technique for quality control and research applications.
Experimental Protocols for Method Comparison
A valid statistical comparison requires that both analytical methods are validated according to international guidelines, such as those from the International Conference on Harmonization (ICH), ensuring that the generated data are reliable and comparable [1] [8]. The following protocols outline the standard approach for developing and validating paired UV and HPLC methods.
Sample Preparation
For both UV and HPLC analysis, a representative sample (e.g., tablet powder) is accurately weighed and dissolved in a suitable solvent, typically methanol or a mobile phase. The solution is then sonicated to ensure complete dissolution, filtered to remove particulate matter, and diluted to a target concentration within the linear range of the respective method [1].
UV Spectroscopy Method
UV analysis is performed using a double-beam UV-Vis spectrophotometer. The wavelength for quantification is selected based on the maximum absorbance of the target analyte (e.g., 241 nm for repaglinide, 234 nm for metformin). Measurements are taken against a solvent blank, and the analyte concentration is determined from a pre-established calibration curve [1] [80].
HPLC-UV Method
HPLC analysis is typically performed using a reversed-phase C18 column. The mobile phase composition is optimized for the specific analyte; for instance, a mixture of methanol and water (80:20 v/v, pH adjusted to 3.5) is used for repaglinide at a flow rate of 1.0 mL/min [1]. Detection is carried out using a UV detector set at the appropriate wavelength. The peak area or height is used for quantification against a calibration curve.
Data Collection for Statistical Testing
To facilitate a paired comparison, multiple samples (n ≥ 6) of the same batch of a pharmaceutical product are prepared and analyzed using both the UV and HPLC methods. The resulting data, such as the measured content of the active ingredient, are recorded in a paired manner for subsequent statistical analysis [8].
Statistical Analysis: t-tests and F-tests
The core of the method agreement study involves applying paired t-tests and F-tests to the data collected from the two techniques.
Application of Paired t-test
A paired t-test is used to compare the accuracy or the mean results obtained from the two methods. It determines if there is a statistically significant difference between the average measured concentrations from UV and HPLC.
- Null Hypothesis (H₀): There is no difference between the mean results of the two methods (μ_diff = 0).
- Alternative Hypothesis (H₁): There is a significant difference between the mean results of the two methods (μ_diff ≠ 0). A p-value greater than the significance level (typically α = 0.05) indicates that there is no statistically significant difference between the means, supporting the agreement in accuracy between the two methods [81].
Application of F-test
An F-test compares the precision (variance) of the two methods. It assesses whether one method is significantly more variable than the other.
- Null Hypothesis (H₀): The variances of the two methods are equal (σ²UV = σ²HPLC).
- Alternative Hypothesis (H₁): The variances of the two methods are not equal (σ²UV ≠ σ²HPLC). A p-value greater than 0.05 suggests that there is no statistically significant difference in the precision of the two methods [81].
Quantitative Data Comparison: UV Spectroscopy vs. HPLC
The following tables synthesize experimental data from multiple studies to provide a comparative overview of the performance of UV and HPLC methods for various pharmaceutical compounds.
Table 1: Comparison of Validation Parameters for UV and HPLC Methods
| Parameter | UV Spectroscopy | HPLC-UV | Compound |
|---|---|---|---|
| Linearity (R²) | > 0.999 [1] | > 0.999 [1] | Repaglinide |
| Precision (% RSD) | < 1.50% [1] | < 1.50% [1] | Repaglinide |
| Precision (% RSD) | 0.59 - 2.12% [8] | 0.83 - 1.58% [8] | Piperine |
| Accuracy (% Recovery) | 99.63 - 100.45% [1] | 99.71 - 100.25% [1] | Repaglinide |
| Accuracy (% Recovery) | 96.7 - 101.5% [8] | 98.2 - 100.6% [8] | Piperine |
| Limit of Detection (LOD) | 0.65 μg/mL [8] | 0.23 μg/mL [8] | Piperine |
| Measurement Uncertainty | 4.29% (at 49.48 g/kg) [8] | 2.47% (at 34.82 g/kg) [8] | Piperine |
Table 2: Performance Comparison for Specific Drug Analyses
| Compound | Method | Key Finding | Reference |
|---|---|---|---|
| Levofloxacin | UV-Vis | Recovery: 96.00 - 99.50% | [18] |
| Levofloxacin | HPLC | Recovery: 96.37 - 110.96% | [18] |
| Metformin | UV-Vis | Recovery: 92 - 104%; RSD: < 3.773% | [80] |
| Metformin | UHPLC | Recovery: 98 - 101%; RSD: < 1.578% | [80] |
| Xylitol | HPLC-UVD | LOD: 0.01 mg/L; Superior for trace analysis | [57] |
Visualization of Statistical Comparison Workflow
The following diagram illustrates the logical workflow for planning, executing, and interpreting a statistical comparison between UV and HPLC methods.
Uncertainty Components in HPLC-UV Analysis
Understanding the sources of uncertainty is crucial for a meaningful method comparison. The following diagram details the key contributors to measurement uncertainty in a typical HPLC-UV analysis, which often has a more complex uncertainty profile than UV spectroscopy.
The Scientist's Toolkit: Essential Research Reagents and Materials
The following table lists key materials and reagents commonly required for the development and validation of UV and HPLC methods in pharmaceutical analysis.
Table 3: Essential Research Reagents and Solutions
| Item | Function | Example Use |
|---|---|---|
| HPLC-Grade Methanol/Acetonitrile | Mobile phase component; solvent for sample preparation. | Used in mobile phase for repaglinide [1] and levetiracetam analysis [76]. |
| Ultra-Pure Water | Mobile phase component; solvent for aqueous samples. | Prevents contamination and baseline noise in HPLC [8]. |
| Reference Standard | Highly pure analyte used to create calibration curves and determine accuracy. | Essential for quantitative method validation [1] [8]. |
| Orthophosphoric Acid | Mobile phase modifier to control pH, improving peak shape and separation. | Used to adjust mobile phase to pH 3.5 for repaglinide analysis [1]. |
| Volumetric Flasks & Pipettes | For precise preparation of standard and sample solutions. | Critical for ensuring accuracy in all quantitative dilutions. |
| Syringe Filters (0.45 μm or 0.22 μm) | Removal of particulate matter from samples prior to injection into the HPLC system. | Prevents column damage and system blockages [76] [57]. |
| C18 Chromatographic Column | Stationary phase for reversed-phase HPLC separation of analytes. | The most common column type; used in multiple cited studies [1] [76]. |
The synthesized experimental data and statistical framework lead to several key conclusions for practitioners:
- HPLC generally offers superior sensitivity and lower measurement uncertainty. This is evidenced by lower LOD/LOQ values and smaller expanded uncertainty budgets, as seen with piperine and xylitol analysis [8] [57]. This makes HPLC the preferred method for trace analysis, stability studies, and when the highest level of accuracy is required.
- UV Spectroscopy is a robust, cost-effective alternative for high-concentration analytes. For quality control of active ingredient content in formulations where the analyte is present in sufficient concentration and the matrix is simple, UV spectroscopy can provide excellent accuracy and precision with faster analysis time and lower operational costs [1].
- Method selection is context-dependent. The choice between UV and HPLC should be guided by the specific application, required sensitivity, complexity of the sample matrix, and available resources. For instance, while UV was adequate for repaglinide tablets, HPLC was necessary for accurately determining levofloxacin release from a complex composite scaffold [18].
In summary, a statistical comparison using t-tests and F-tests provides a scientifically rigorous foundation for selecting an analytical method. While HPLC often demonstrates superior performance metrics, UV spectroscopy remains a highly valuable and reliable technique within its operational domain.
The selection of an appropriate analytical method is a critical decision in pharmaceutical development, influencing everything from routine quality control to regulatory submission success. Within the framework of a broader research thesis comparing the measurement uncertainty of UV spectroscopy and HPLC, understanding the regulatory landscape is paramount. The International Council for Harmonisation (ICH), the United States Pharmacopeia (USP), and the European Pharmacopoeia (EP) provide the essential frameworks for validating these methods and demonstrating their suitability [82]. While these guidelines are highly harmonized, subtle differences in their application can impact method selection, validation protocols, and ultimately, regulatory acceptance. This guide provides an objective comparison of UV spectroscopy and HPLC through the lens of these regulatory standards, supported by experimental data and structured to inform decision-making for drug development professionals.
Regulatory Framework: ICH, USP, and EP
The ICH guideline Q2(R1), "Validation of Analytical Procedures: Text and Methodology," serves as the foundational document for analytical method validation globally [82]. It outlines the core validation parameters—including accuracy, precision, specificity, linearity, and range—required to prove a method is suitable for its intended purpose.
The USP and EP have integrated these principles into their respective general chapters:
- USP General Chapter <1225>, "Validation of Compendial Procedures," aligns closely with ICH Q2(R1) but places a stronger emphasis on system suitability testing (SST) as a prerequisite for method validation [82].
- EP General Chapter 5.15, "Validation of Analytical Procedures," fully adopts ICH Q2(R1) but provides supplementary guidance on specific analytical techniques, with a particular focus on robustness for methods used in stability studies [82].
A key similarity is that all three bodies adopt a risk-based approach, allowing flexibility in the extent of validation based on the method's intended use [82]. The most notable difference in terminology is that the USP uses the term "ruggedness" in a context similar to the ICH's "intermediate precision" [82].
Table 1: Key Regulatory Guidelines for Analytical Method Validation
| Regulatory Body | Primary Guideline | Key Focus Areas | Terminology Notes |
|---|---|---|---|
| ICH | Q2(R1) | Science and risk-based approach; global foundation | "Intermediate precision" |
| USP | General Chapter <1225> | System suitability testing (SST); compendial methods | "Ruggedness" |
| European Pharmacopoeia (EP) | General Chapter 5.15 | Robustness; specific analytical techniques; stability | Fully aligned with ICH Q2(R1) |
Comparative Analysis: UV Spectroscopy vs. HPLC
The choice between UV spectroscopy and HPLC is fundamentally dictated by the analytical problem's complexity and regulatory requirements.
Principle and Specificity
- UV Spectroscopy: This technique operates on the Beer-Lambert Law, measuring the absorption of ultraviolet light by a sample at a specific wavelength [68]. It is a non-separative technique. Consequently, it has low specificity if excipients, impurities, or degradation products contain chromophores that absorb at the same wavelength as the analyte, leading to potential interference [68].
- HPLC: This is a separative technique where compounds are distributed between a stationary phase and a mobile phase [68]. Detection, often via UV, occurs after separation. This provides high specificity, as each compound has a distinct retention time, allowing the target analyte to be resolved from impurities, excipients, and degradation products [68].
Regulatory Acceptance and Applications
The specificity of a method directly influences its regulatory acceptance for various applications.
- UV Spectroscopy Applications: It is suitable for routine assay of raw materials and single-active-ingredient formulations, dissolution testing (when free from interference), and quick sample screening [68]. However, it is rarely accepted as the sole method for complex formulations or regulatory submissions unless a lack of interference is conclusively demonstrated [68].
- HPLC Applications: HPLC is considered the gold standard for pharmacopeial methods [68]. It is the preferred technique for drug assay and impurity profiling, stability-indicating methods, bioequivalence studies, and analysis of multicomponent formulations [68]. It is widely accepted by regulatory bodies (FDA, EMA, etc.) for submission dossiers [82] [68].
Table 2: Direct Comparison of UV Spectroscopy and HPLC
| Characteristic | UV Spectroscopy | High-Performance Liquid Chromatography (HPLC) |
|---|---|---|
| Principle | Beer-Lambert Law (Absorbance measurement) [68] | Separation based on partitioning [68] |
| Specificity | Low; susceptible to interference [68] | High; separates analytes from interference [68] |
| Sensitivity | Moderate (usually µg/mL) [68] | High (UV: ng-µg/mL; MS: pg-ng/mL) [68] |
| Analysis Time | Fast (minutes) [68] | Longer (10-60 minutes per run) [68] |
| Cost | Low (instrument and solvents) [68] | High (columns, solvents, maintenance) [68] |
| Ideal For | Single-component analysis, quick QC checks [68] | Multicomponent mixtures, impurities, stability studies [68] |
Experimental Data and Method Validation
The following experimental data, derived from published studies, illustrates the typical validation outcomes for UV and HPLC methods and provides context for their measurement uncertainty.
Case Study: Analysis of Repaglinide
A study developed and validated both UV and HPLC methods for the antidiabetic drug repaglinide in tablets, following ICH Q2(R1) guidelines [1].
- UV-Spectrophotometric Method: The analyte was dissolved in methanol and measured at 241 nm. The method demonstrated excellent linearity (r² > 0.999) in the range of 5-30 μg/mL. The mean recovery was 99.63-100.45%, and the precision (RSD) was below 1.5% [1].
- HPLC Method: Analysis used a reversed-phase C18 column with a mobile phase of methanol:water (80:20, v/v, pH 3.5) at a flow rate of 1.0 mL/min. Detection was at 241 nm. The method was linear (r² > 0.999) from 5-50 μg/mL. The mean recovery was 99.71-100.25%, with precision (RSD) values lower than those of the UV method [1].
Table 3: Validation Data for Repaglinide Analysis [1]
| Validation Parameter | UV-Spectrophotometric Method | Reversed-Phase HPLC Method |
|---|---|---|
| Linearity Range | 5 - 30 μg/mL | 5 - 50 μg/mL |
| Correlation Coefficient (r²) | > 0.999 | > 0.999 |
| Precision (% RSD) | < 1.5% | More precise than UV |
| Accuracy (% Recovery) | 99.63 - 100.45% | 99.71 - 100.25% |
Case Study: Analysis of Lamivudine
Another comparative study for the antiviral drug lamivudine also validated UV, RP-HPLC, and HPTLC methods per ICH Q2(R1) [83].
- The RP-HPLC method was concluded to be superior for the quantification of lamivudine in tablet formulation due to its high reproducibility, good retention time, sensitivity, and higher percent recovery [83]. The method also proved to be stability-indicating, successfully separating the drug peak from degradation products formed under forced degradation conditions [83].
Measurement Uncertainty in HPLC and UV
Understanding the sources and propagation of uncertainty is a core component of method suitability.
Research on the propagation of uncertainty in HPLC with UV-VIS detection identifies that the uncertainty in the eluate concentration at the detector is not zero, even if the injected standard concentration is known exactly [5]. The analysis reveals that among instrumental factors, the uncertainty in injection volume contributes the greatest uncertainty to the final result [5]. Furthermore, studies suggest that the relative uncertainty of a measured peak height tends to be greater than that of a peak area [5].
A 2024 study comparing HPLC detectors for xylitol analysis provided quantitative uncertainty data, reporting that a well-optimized HPLC-UV method exhibited a low relative expanded uncertainty in the range of 1.12–3.98% [57]. This metric is crucial for understanding the reliability of quantitative results.
The Scientist's Toolkit: Essential Research Reagents and Materials
The following table lists key materials and reagents commonly used in the development and validation of UV and HPLC methods, based on the protocols cited [1] [57] [83].
Table 4: Key Research Reagent Solutions for Method Development
| Item | Function in Analysis | Example from Literature |
|---|---|---|
| HPLC-Grade Methanol / Acetonitrile | Mobile phase component; solvent for standard and sample preparation [1] [83]. | Used in mobile phase for repaglinide (MeOH:Water) [1] and lamivudine (MeOH:Water) [83]. |
| HPLC-Grade Water | Mobile phase component [1] [83]. | Used in mobile phases for repaglinide and lamivudine analysis [1] [83]. |
| Buffers / pH Adjustors | Modifies mobile phase to control ionization, improve peak shape, and enhance separation. | Orthophosphoric acid used to adjust mobile phase to pH 3.5 for repaglinide analysis [1]. |
| Derivatization Reagent | Reacts with non-UV-absorbing analytes to produce a UV-absorbing derivative for detection. | p-Nitrobenzoyl chloride (PNBC) used to derivative xylitol for HPLC-UVD analysis [57]. |
| Reference Standard | Highly characterized substance used to prepare calibration standards for quantitative analysis. | Repaglinide reference standard from USV Lab. Pvt. Ltd. [1]; Lamivudine from Cipla Ltd [83]. |
Workflow and Decision Pathway
The following diagram illustrates the logical relationship between the core principles of the analytical techniques, the resulting data characteristics, and the subsequent regulatory implications, as discussed in the comparative analysis.
The selection between UV spectroscopy and HPLC is a strategic decision balanced against analytical needs, operational resources, and regulatory requirements. UV spectroscopy offers a fast, simple, and cost-effective solution for specific, well-defined applications such as raw material identification or single-component assay where interference is absent. However, its low specificity limits its use in complex scenarios.
HPLC, with its superior separation power, provides the high specificity, sensitivity, and robustness demanded for most regulatory submissions. It is the unequivocal choice for impurity profiling, stability-indicating methods, and analysis of multicomponent formulations. While more costly and time-intensive, its comprehensive data and alignment with pharmacopeial standards make it the gold standard. Ultimately, demonstrating method suitability through rigorous validation as per ICH, USP, and EP guidelines—with a clear understanding of the inherent measurement uncertainties—is the critical factor for regulatory acceptance, regardless of the technique chosen.
In pharmaceutical research and drug development, the selection of an analytical technique is a critical decision that hinges on a fundamental trade-off: the need for precise, reliable data versus the constraints of budget, time, and operational complexity. High-performance liquid chromatography (HPLC) and ultraviolet-visible spectroscopy (UV-Vis) represent two cornerstone methodologies in the analyst's toolkit, each with distinct advantages and limitations. This guide provides an objective comparison of these techniques, focusing on their performance characteristics, operational requirements, and optimal application domains. The data and experimental protocols presented herein are drawn from contemporary scientific literature, enabling professionals to make informed decisions aligned with their specific precision requirements and resource constraints.
HPLC is presently considered one of the most successful separation methods because it is flexible, fast, accurate, and gentle, destroying none but the most labile compounds [84]. UV detectors are undoubtedly the most frequently used type of detector with HPLC systems, and this will continue for many years, despite the rise of the modular mass spectrometric detector [85]. Conversely, UV-Vis spectrophotometry offers a simpler, more accessible approach for certain applications, though with generally reduced specificity. Understanding the precise performance differentials between these techniques is essential for effective resource allocation in research and quality control environments.
Technical Performance Comparison
Quantitative Analysis of Measurement Performance
Direct comparative studies provide the most reliable evidence for evaluating the performance of HPLC and UV-Vis techniques. The following table summarizes key validation parameters from research that simultaneously applied both methods to the same analytical problems.
Table 1: Direct Comparison of HPLC and UV-Vis Performance in Pharmaceutical Analysis
| Analyte/Study | Technique | Accuracy (%) | Precision (RSD%) | Linearity (R²) | LOD/LOQ | Measurement Uncertainty |
|---|---|---|---|---|---|---|
| Piperine in Black Pepper [8] | HPLC-UV | 98.2 – 100.6 | 0.83 – 1.58 | Good | LOD: 0.23 | 2.47% (at 34.819 g/kg) |
| UV Spectroscopy | 96.7 – 101.5 | 0.59 – 2.12 | Good | LOD: 0.65 | 4.29% (at 49.481 g/kg) | |
| Levofloxacin in Scaffolds [18] | HPLC | 96.37 – 110.96 | N/R | 0.9991 | N/R | N/R |
| UV-Vis | 96.00 – 99.50 | N/R | 0.9999 | N/R | N/R | |
| Metformin HCl in Tablets [80] | UHPLC | 98 – 101 | Repeatability: <1.578% | Within 2.5–40 μg/ml | LLOQ: 0.625 μg/ml | N/R |
| UV-Vis | 92 – 104 | Repeatability: <3.773% | Within 2.5–40 μg/ml | LLOD: 0.156 μg/ml | N/R | |
| Methamphetamine [30] | HPLC-UV | N/R | N/R | N/R | N/R | RMSE: 1.1 |
| Benchtop NMR | N/R | N/R | N/R | N/R | RMSE: 2.1 |
(N/R: Not Reported in the cited study)
The data consistently demonstrates that HPLC offers superior specificity and accuracy, particularly in complex matrices. For piperine analysis, HPLC showed lower measurement uncertainty (2.47%) compared to UV spectroscopy (4.29%) [8]. Similarly, for metformin hydrochloride, the UHPLC method demonstrated both higher accuracy (98-101% vs. 92-104%) and better precision (RSD <1.578% vs. <3.773%) compared to the UV-Vis method [80].
General Performance Characteristics and Limitations
Beyond direct comparisons, understanding the inherent capabilities and limitations of each technique is crucial for method selection.
Table 2: General Performance Characteristics of HPLC-UV and UV-Vis Spectrophotometry
| Performance Characteristic | HPLC-UV | UV-Vis Spectrophotometry |
|---|---|---|
| Specificity | High (separates components) | Low to Moderate (measures total absorbance) |
| Typical API Assay Precision (RSD%) | 0.6 - 1.1% [86] | Varies widely (often >2%) |
| Process Capability (for API assays) | ~3σ [86] | Not typically used for API potency |
| Sensitivity | Excellent (depends on detector) | Good |
| Matrix Tolerance | Moderate to High (separation helps) | Low (susceptible to interference) |
| Key Limitations | Higher cost, complexity, method development time | Lack of specificity for mixtures, solvent interference at low wavelengths [85] |
A critical consideration for HPLC in regulated environments is its variability. The cumulative intermediate precision for HPLC assays of Active Pharmaceutical Ingredients (APIs) is approximately 1.1% RSD [86]. This variability can impact the ability to trend data effectively and may lead to "false out-of-specification" results, particularly when monitoring stable drug substances over time [86].
Experimental Protocols from Comparative Studies
Protocol 1: Determination of Piperine in Black Pepper
This protocol demonstrates a direct comparison of performance parameters and measurement uncertainty.
- Objective: To compare the validation parameters and measurement uncertainties of UV spectroscopy and HPLC-UV for the determination of piperine in black pepper [8].
- Sample Preparation: Black pepper samples were ground using a blender and sieved through a 60-mesh. For analysis, standards were fortified in selected samples [8].
- UV Spectroscopy Method: Specificity was evaluated by comparing spectra. The limit of detection (LOD) was determined using the formula LOD = (3.3 × σ)/S, where σ is the standard deviation of the intercept and S is the average slope of the calibration curve. Accuracy and precision were confirmed by analyzing 18 samples fortified with 0.5, 2, and 5% piperine [8].
- HPLC-UV Method: Specificity was defined as the isolation of a piperine peak free from interference, identified by comparing retention times with reference standards. The LOD was calculated similarly to the UV method. Accuracy and precision were assessed over the same fortification levels [8].
- Measurement Uncertainty: Estimated according to NIST and Eurachem guidelines by combining all uncertainties from selected sources, including type A (statistical calculations) and type B (given expanded uncertainties) standard uncertainties [8].
Protocol 2: Analysis of Levofloxacin Released from Composite Scaffolds
This protocol highlights the importance of technique selection in complex drug-delivery systems.
- Objective: To compare HPLC and UV-Vis for determining Levofloxacin released from mesoporous silica microspheres/nano-hydroxyapatite composite scaffolds [18].
- HPLC Conditions:
- Instrument: Shimadzu liquid chromatograph with LC-2010AHT pump and UV-Vis detector.
- Column: Sepax BR-C18 (250 × 4.6 mm, 5 μm).
- Mobile Phase: 0.01 mol/L KH₂PO₄, methanol, and 0.5 mol/L tetrabutylammonium hydrogen sulphate (75:25:4).
- Flow Rate: 1 mL/min.
- Detection: 290 nm.
- Internal Standard: Ciprofloxacin [18].
- UV-Vis Method:
- Instrument: UV-2600 UV-Vis spectrophotometer.
- Wavelength Selection: Standard solutions at high (50 μg/mL), medium (25 μg/mL), and low (5 μg/mL) concentrations were scanned from 200–400 nm to determine maximum absorption.
- Sample Preparation for Both Methods: Standard solutions of Levofloxacin were prepared in simulated body fluid across 14 concentration gradients (300 - 0.01 μg/mL). For HPLC, samples involved liquid-liquid extraction with dichloromethane and drying under nitrogen [18].
Decision Framework and Workflow Visualization
The choice between HPLC and UV-Vis is not merely a technical preference but a strategic decision impacting data quality, operational costs, and project timelines. The following diagram illustrates a logical workflow for selecting the appropriate technique based on analytical requirements and sample characteristics.
Figure 1: Analytical Technique Selection Workflow
Resource Considerations and Operational Costs
Cost and Infrastructure Implications
Beyond technical performance, the economic and operational aspects of analytical techniques significantly influence their suitability for different laboratory environments.
- Capital Investment: A basic HPLC-UV system represents a substantial capital investment, often ranging from tens to hundreds of thousands of dollars depending on configuration and capabilities. In comparison, a stand-alone UV-Vis spectrophotometer is significantly less expensive, typically costing a few thousand to tens of thousands of dollars [87].
- Consumables and Solvents: HPLC operations require high-purity solvents, columns, and other consumables that contribute to ongoing operational costs. The method relies heavily on toxic and/or expensive solvents, which complicate its application and increase operational costs [30]. UV-Vis methods generally have lower consumable costs, primarily limited to cuvettes and reagents.
- Maintenance and Expertise: HPLC systems require regular maintenance, qualified operators, and method development expertise. The typical error attributable to a drug substance HPLC assay method is about 1%, partly due to this operational complexity [86]. UV-Vis systems are generally simpler to operate and maintain, requiring less specialized training.
- Throughput and Time Considerations: For simple analyses, UV-Vis typically offers higher throughput as it often requires minimal sample preparation and no separation step. HPLC, while slower per sample, provides multiplexed information (separation and quantification of multiple components) in a single run.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential Materials for HPLC-UV and UV-Vis Analyses
| Item | Function | Application Notes |
|---|---|---|
| HPLC-Grade Solvents | Mobile phase components | Essential for HPLC to avoid system damage and baseline noise; acetonitrile and methanol are most common [18]. |
| Buffer Salts | Mobile phase modifiers | Control pH and ionic strength; phosphate and acetate buffers are widely used [88]. |
| HPLC Columns | Stationary phase for separation | C18 columns are most common; choice depends on analyte properties [88]. |
| UV-Vis Cuvettes | Sample containers for measurement | Must have appropriate transmission characteristics for selected wavelengths. |
| Reference Standards | Calibration and quantification | High-purity characterized materials essential for both techniques [8]. |
| Syringe Filters | Sample clarification | Remove particulates that could damage HPLC systems or scatter light in UV-Vis [18]. |
The choice between HPLC and UV-Vis spectrophotometry represents a classic trade-off between analytical performance and resource allocation. HPLC-UV provides superior specificity, accuracy, and lower measurement uncertainty, making it the unequivocal choice for regulatory submissions, complex matrices, and situations requiring definitive identification and quantification of multiple components. However, this capability comes with significantly higher capital and operational costs, greater complexity, and longer method development times.
UV-Vis spectrophotometry offers a cost-effective, rapid, and operationally simpler alternative for applications where its limitations are not prohibitive. It remains fit-for-purpose for routine analysis of pure substances, educational applications, and situations where budgetary constraints are primary and the technique's lower specificity is acceptable.
In an era of increasing analytical sophistication, the decision framework presented here empowers scientists to make judicious, evidence-based selections that balance precision requirements with practical resource constraints, ultimately optimizing laboratory efficiency and data quality in pharmaceutical research and development.
Selecting the optimal analytical technique is a fundamental challenge in research and drug development. The choice between seemingly straightforward methods like Ultraviolet-Visible (UV) spectroscopy and more complex ones like High-Performance Liquid Chromatography (HPLC), or deciding when to use them together, has significant implications for data reliability, efficiency, and cost. Ultraviolet-Visible (UV) spectroscopy and High-Performance Liquid Chromatography (HPLC) represent different classes of analytical techniques: one provides a rapid, composite profile of a sample, while the other separates and individually quantifies its components. The decision is not always straightforward and must be guided by the sample's complexity, the information required, and the required standard of proof. This guide provides a structured framework for this decision-making process, grounded in experimental data and a rigorous comparison of performance characteristics, including the critical parameter of measurement uncertainty.
Fundamental Principles
UV Spectroscopy: This technique measures the absorption of ultraviolet or visible light by a sample at specific wavelengths. The absorbance follows the Beer-Lambert Law, where absorbance is proportional to the concentration of the absorbing species and the path length [89]. It provides a single, composite measurement for all chromophoric compounds in the solution.
HPLC-UV: This is a hybrid technique that combines separation and detection. The HPLC system separates components in a mixture based on their interaction with a stationary phase and a mobile phase. The separated analytes then pass through a UV flow cell, where they are detected based on their UV absorbance [89] [90]. It provides individual quantitative data for each separated component.
Key Technical Characteristics
The table below summarizes the core attributes of each technique.
Table 1: Fundamental Characteristics of UV Spectroscopy and HPLC-UV
| Characteristic | UV Spectroscopy | HPLC-UV |
|---|---|---|
| Principle | Absorption of light by chromophores [89] | Separation followed by UV detection [89] |
| Information Provided | Total chromophore content; composite spectrum | Individual quantification of separated components |
| Analysis Speed | Very fast (seconds to minutes) | Slower (minutes to tens of minutes) |
| Sample Requirements | Must contain a UV chromophore | Must contain a UV chromophore and be soluble in the mobile phase [91] |
| Key Strength | Simplicity, speed, and cost-effectiveness | Selectivity, specificity, and ability to handle complex mixtures |
Comparative Experimental Data: Performance and Reliability
Direct comparisons in scientific literature highlight the practical performance differences between these methods.
Case Study 1: Pharmaceutical Analysis of Repaglinide
A study developing methods for the antidiabetic drug repaglinide in tablets demonstrated the performance of both techniques when optimally applied to a simple mixture (a drug in a formulated tablet) [1].
Table 2: Method Validation Parameters for Repaglinide Analysis [1]
| Validation Parameter | UV Spectrophotometry | HPLC Method |
|---|---|---|
| Linearity Range | 5–30 μg/mL | 5–50 μg/mL |
| Correlation Coefficient (r²) | >0.999 | >0.999 |
| Precision (% R.S.D.) | <1.50 | Better than UV method |
| Mean Recovery | 99.63–100.45% | 99.71–100.25% |
Conclusion: For this relatively simple analysis of a single active ingredient in a formulation, both methods demonstrated excellent and comparable accuracy and linearity. However, the HPLC method showed superior precision and a wider linear range [1].
Case Study 2: Drug Delivery System Analysis
A more complex scenario involved quantifying Levofloxacin released from a novel composite scaffold (mesoporous silica microspheres/nano-hydroxyapatite). In this case, the sample matrix was complex, containing multiple components that could interfere with analysis [18].
Table 3: Recovery Rates for Levofloxacin from a Complex Scaffold [18]
| Method | Low Concentration Recovery | Medium Concentration Recovery | High Concentration Recovery |
|---|---|---|---|
| HPLC | 96.37 ± 0.50% | 110.96 ± 0.23% | 104.79 ± 0.06% |
| UV-Vis | 96.00 ± 2.00% | 99.50 ± 0.00% | 98.67 ± 0.06% |
Conclusion: The study found that UV-Vis was not accurate for measuring drug concentration in this complex, multi-component system due to interference from the scaffold's components. HPLC was determined to be the preferred method due to its superior selectivity, which allowed for accurate quantification of Levofloxacin amidst potential interferents [18].
Measurement Uncertainty: A Critical Differentiator
Measurement uncertainty (MU) is a paramount parameter that quantifies the reliability and precision of an analytical result, which is especially critical for regulatory compliance and quality control [92] [14].
Uncertainty in HPLC-UV Methods
Studies quantifying MU for HPLC-UV methods identify the primary sources of variability. An analysis of Metopimazine determination found that sample volume and calibration standard concentration were the dominant contributors, accounting for over 76% of the total uncertainty [92]. Another study on organic acids in food reported an expanded uncertainty (at a 95% confidence level) ranging from 2.79% to 8.92% for various analytes [14]. This level of rigorous uncertainty estimation is a standard expectation for HPLC methods in regulated environments.
Comparative Uncertainty of UV vs. HPLC
UV spectroscopy, while precise, can have higher uncertainty in complex samples due to its lack of selectivity. Any change in the composite absorbance caused by interferents, sample turbidity, or instrumental drift translates directly into a concentration error for the target analyte. In contrast, HPLC's separation step minimizes the impact of these interferents, typically leading to lower measurement uncertainty for mixtures, albeit with a more complex uncertainty budget originating from the chromatographic process itself [92] [93].
Decision Framework: Selecting the Right Technique
The following workflow provides a systematic approach to selecting an analytical technique based on sample characteristics and analytical requirements.
Diagram 1: Analytical Technique Selection Workflow
Key Decision Factors
For UV Spectroscopy: Opt for this method when analyzing pure compounds or simple mixtures with known spectral characteristics, when the target analytes possess strong chromophores, and when the analysis requires high throughput and low cost. It is ideal for routine quality control of raw materials or finished products where interference is minimal [1].
For HPLC-UV: Choose this technique for complex mixtures, when the method requires selectivity and specificity to resolve multiple components, or when you need to confirm the identity and purity of a peak (e.g., using a Photodiode Array detector for spectral confirmation) [18] [90]. It is indispensable for stability-indicating methods and assays requiring low measurement uncertainty.
For Complementary and Advanced Techniques: In research and development, techniques are often used together. HPLC can separate components for quantification, while Nuclear Magnetic Resonance (NMR) can be used to identify unknown impurities or confirm structures [91]. For compounds without a chromophore (e.g., sugars), universal detectors like Refractive Index (RI) or Charged Aerosol Detectors (CAD) are coupled with HPLC [94].
The Scientist's Toolkit: Essential Reagents and Materials
Table 4: Key Research Reagent Solutions and Their Functions
| Reagent/Material | Function in Analysis | Example Use Case |
|---|---|---|
| HPLC-Grade Solvents | Serve as the mobile phase; high purity is critical to minimize baseline noise and ghost peaks. | Acetonitrile and methanol are used for reversed-phase HPLC [1] [95]. |
| Buffers & Additives | Modify the mobile phase pH and ionic strength to control separation, peak shape, and retention times. | Ammonium acetate/formate buffers; orthophosphoric acid for pH adjustment [1] [92]. |
| Certified Reference Standards | Used for instrument calibration and method validation; their purity and traceability are essential for accurate quantification and low measurement uncertainty [92]. | Determining the concentration of an active pharmaceutical ingredient (API) [1]. |
| Solid Phase Extraction Kits | Clean up and pre-concentrate samples before analysis, reducing matrix effects and improving sensitivity. | Modified QuEChERS method for pesticide residue analysis in food [93]. |
| Chromatography Columns | The heart of the HPLC system where chemical separation occurs; selection (C8, C18, etc.) is based on analyte properties. | C18 columns are common for reversed-phase separation of small molecules [1] [18]. |
There is no single "best" technique for every scenario. The choice between UV spectroscopy and HPLC-UV is a trade-off between speed and simplicity versus selectivity and specificity. UV spectroscopy offers an efficient and cost-effective solution for simple, well-defined analyses. In contrast, HPLC-UV is the unequivocal choice for complex mixtures, providing the selectivity needed for accurate quantification and purity assessment, which directly translates to lower measurement uncertainty and higher data reliability. By applying the structured framework and considering the experimental data presented, scientists and drug development professionals can make informed, justified decisions that ensure the integrity of their analytical results.
Conclusion
The comparison between UV spectroscopy and HPLC reveals a clear trade-off between simplicity and specificity, with direct implications for measurement uncertainty. UV spectroscopy offers rapid, cost-effective analysis for well-characterized, single-component systems but demonstrates higher uncertainty with complex mixtures due to limited specificity. HPLC provides superior separation capabilities and lower uncertainty for multicomponent analysis, impurity profiling, and regulatory submissions, though with increased operational complexity and cost. The choice between techniques should be guided by fitness-for-purpose, considering the analytical requirements, sample complexity, and regulatory context. Future directions point toward integrated approaches, combining rapid screening with confirmatory analysis, and the adoption of advanced data processing techniques like Quantum Mechanical Modeling to further reduce uncertainty in pharmaceutical analysis and clinical research.
References
- 1. Comparison of UV spectrophotometry and high ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3658086/]
- 2. Validation of Stability-Indicating HPLC Methods for ... [https://www.chromatographyonline.com/view/validation-of-stability-indicating-hplc-methods-for-pharmaceuticals-overview-methodologies-and-case-studies]
- 3. Estimating Uncertainty [https://www.chromatographyonline.com/view/estimating-uncertainty-0]
- 4. Uncertainty sources in UV-Vis spectrophotometric ... [https://link.springer.com/article/10.1007/s00769-006-0124-x]
- 5. Propagation of uncertainty in high-performance liquid ... [https://www.sciencedirect.com/science/article/abs/pii/S0003267001012223]
- 6. Comparison of Quantification Using UV-Vis, NMR, and ... [https://www.mdpi.com/1422-0067/26/14/6638]
- 7. Development and validation of RP-HPLC and UV ... [https://arabjchem.org/development-and-validation-of-rp-hplc-and-uv-spectrophotometric-methods-for-rapid-simultaneous-estimation-of-amlodipine-and-benazepril-in-pure-and-fixed-dose-combination/]
- 8. Comparison study of validation parameters and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9339058/]
- 9. UV-Vis Spectroscopy: Principle, Strengths and Limitations ... [https://www.technologynetworks.com/analysis/articles/uv-vis-spectroscopy-principle-strengths-and-limitations-and-applications-349865]
- 10. Comparative Study of UV And HPLC Methods for ... [https://www.ijsrtjournal.com/article/Comparative-Study-of-UV-And-HPLC-Methods-for-Estimation-of-Drug]
- 11. Spectrophotometry UV : Principles, Applications... | LinkedIn vs HPLC [https://www.linkedin.com/posts/abedin-shohagh_uv-spectrophotometry-vs-hplc-1-principle-activity-7373754535925833728--AST]
- 12. Quantitative evaluation of true-to-life nanoplastics using UV -visible... [https://pubs.rsc.org/en/content/articlehtml/2025/en/d5en00502g]
- 13. Errors in Spectrophotometry and Calibration Procedures to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5293527/]
- 14. Validation, measurement uncertainty, and application of ... [https://www.sciencedirect.com/science/article/abs/pii/S0889157525001498]
- 15. Open-access Measurement Uncertainty Calculator MUCalc ... [https://www.sciencedirect.com/science/article/pii/S1355030624001060]
- 16. Peak Integration Errors: Common Issues and How to Fix ... [https://www.sepscience.com/hplc-solutions-129-peak-integration-part-3common-integration-errors-6962]
- 17. A Dual Perspective on Bottom-up and Top-Down Approaches [https://pubmed.ncbi.nlm.nih.gov/41126565/]
- 18. Comparison of high-performance liquid chromatography and ultraviolet-visible spectrophotometry to determine the best method to assess Levofloxacin released from mesoporous silica microspheres/nano-hydroxyapatite composite scaffolds - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6425260/]
- 19. Development of an HPLC-UV Method for the Analysis of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3619551/]
- 20. UV Spectroscopy Market Size & Share Analysis [https://www.mordorintelligence.com/industry-reports/uv-spectroscopy-market]
- 21. Analytical High Performance Liquid Chromatograph (HPLC) [https://www.archivemarketresearch.com/reports/analytical-high-performance-liquid-chromatograph-hplc-490357]
- 22. Validation and measurement uncertainty of HPLC-UV ... [https://koreascience.kr/article/JAKO202105558294280.page]
- 23. Data Science Tools For the Prediction of VUV Spectra [https://www.chromatographyonline.com/view/data-science-tools-for-the-prediction-of-vuv-spectra-an-hplc-2025-video-interview-with-kevin-schug]
- 24. Measurement uncertainty [https://en.wikipedia.org/wiki/Measurement_uncertainty]
- 25. Type A and Type B Uncertainty: Evaluating ... [https://www.isobudgets.com/type-a-and-type-b-uncertainty/]
- 26. Random vs. Systematic Error [https://www.physics.umd.edu/courses/Phys276/Hill/Information/Notes/ErrorAnalysis.html]
- 27. Evaluating uncertainty components: Type B [https://physics.nist.gov/cuu/Uncertainty/typeb.html]
- 28. Random vs. Systematic Error | Definition & Examples [https://www.scribbr.com/methodology/random-vs-systematic-error/]
- 29. Comparison of HPLC and UV Spectrophotometric Methods ... [https://brieflands.com/journals/ijpr/articles/124310]
- 30. Comparative Analysis of Benchtop NMR and HPLC‐UV for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12500361/]
- 31. difference between UV-spectrophotometer and HPLC- ... [http://www.chromforum.org/viewtopic.php?t=10576]
- 32. UV Spectrophotometry as a Pharmaceutical Testing Solution [https://www.hunterlab.com/blog/uv-spectrophotometry-as-a-pharmaceutical-testing-solution/]
- 33. single component.pptx [https://www.slideshare.net/slideshow/single-componentpptx/254985217]
- 34. From Detection to Imaging: UV Spectroscopy for ... [https://www.americanpharmaceuticalreview.com/Featured-Articles/177714-From-Detection-to-Imaging-UV-Spectroscopy-for-Dissolution-in-Pharmaceutical-Development/]
- 35. RMID - Raw Material Identification [https://bwtek.com/applications/raw-material-id/]
- 36. Application of UV dissolution imaging to pharmaceutical ... [https://www.sciencedirect.com/science/article/abs/pii/S0169409X21003422]
- 37. Impurity profiling HPLC–UV/PDA method development ... [https://link.springer.com/article/10.1007/s44371-025-00366-x]
- 38. A new RP-HPLC approach for estimation of potential ... [https://bmcpharmacoltoxicol.biomedcentral.com/articles/10.1186/s40360-025-00892-5]
- 39. Optimization of HPLC methods for the development ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12105191/]
- 40. Development and validation of a RP-HPLC method for ... [https://www.nature.com/articles/s41598-025-09904-0]
- 41. Development and validation of a RP-HPLC method for the ... [https://link.springer.com/article/10.1007/s44371-025-00293-x]
- 42. A Well-Written Analytical Procedure for Regulated HPLC ... [https://www.chromatographyonline.com/view/a-well-written-analytical-procedure-for-regulated-hplc-testing]
- 43. Comparative validation of UV-spectrophotometry and RP ... [https://pubmed.ncbi.nlm.nih.gov/39603316/]
- 44. Comparative validation of UV-spectrophotometry and RP ... [https://www.sciencedirect.com/science/article/abs/pii/S0003269724002689]
- 45. Risk of false pharmaceutical equivalence (non ... [https://www.sciencedirect.com/science/article/abs/pii/S0731708521003800]
- 46. Development and Validation of Spectrophotometric, Atomic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3239224/]
- 47. Determination of Cefixime by a Validated Stability- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3700077/]
- 48. Optimization of HPLC method for determination of cefixime ... [https://www.sciencedirect.com/science/article/pii/S1319016415000171]
- 49. UV Spectroscopy Gains Use in Dissolution Testing [https://www.pharmtech.com/view/uv-spectroscopy-gains-use-dissolution-testing]
- 50. Comparative Study of RP-HPLC and UV Spectrophotometric ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3021696/]
- 51. Measurement uncertainty arising from sampling and ... [https://pubmed.ncbi.nlm.nih.gov/37348366/]
- 52. Measurement uncertainty arising from sampling and ... [https://www.sciencedirect.com/science/article/abs/pii/S0026265X23011712]
- 53. Validation, measurement uncertainty, and application of ... [https://www.sciencedirect.com/science/article/pii/S0889157525001498]
- 54. Evaluation of the measurement uncertainty: Some common ... [https://www.sciencedirect.com/science/article/abs/pii/S0021967317304909]
- 55. Development and Validation of a HPLC and an UV ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3263711/]
- 56. Development and validation of a novel HPLC-UV method ... [https://www.sciencedirect.com/science/article/pii/S2405844023010770]
- 57. Comparison of three HPLC analytical methods: ELSD, RID ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11364729/]
- 58. How to overcome interferences in UV – Visible ... [https://lab-training.com/overcome-interferences-uv-visible-spectroscopic-studies/]
- 59. A DoE-guided development and validation of a novel ... [https://pubs.rsc.org/en/content/articlelanding/2025/ay/d5ay00310e]
- 60. HPLC Update Day 2: Modern Method Development [https://www.chromatographyonline.com/view/hplc-update-day-2-modern-method-development]
- 61. Artificial Intelligence in HPLC Method Development [https://pubmed.ncbi.nlm.nih.gov/41146636/]
- 62. The Best HPLC Systems: A Buyer's Review of Price and ... [https://www.labx.com/resources/the-best-hplc-systems-a-buyers-review-of-price-and-features/4819]
- 63. Comparative Study of UV And HPLC Methods for ... [https://www.ijsrtjournal.com/article/Comparative+Study+of+UV+And+HPLC+Methods+for+Estimation+of+Drug]
- 64. HPLC Method Development and Validation for ... [https://www.pharmtech.com/view/hplc-method-development-and-validation-pharmaceutical-analysis]
- 65. Mastering HPLC: A Comprehensive Guide to Types and ... [https://www.gmi-inc.com/mastering-hplc-a-comprehensive-guide-to-types-and-their-applications/]
- 66. Development and validation of a new HPLC analytical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6323142/]
- 67. A Simple Approach to Performance Optimization in HPLC ... [https://www.chromatographyonline.com/view/simple-approach-performance-optimization-hplc-and-its-application-ultrafast-separation-development]
- 68. UV Spectrophotometry vs HPLC: Key Differences [https://www.linkedin.com/posts/satish-s-mane_uv-spectrophotometry-vs-hplc-1-principle-activity-7374455512681086976-wOHu]
- 69. Head-to-Head Comparison of High-Performance Liquid ... [https://www.mdpi.com/1420-3049/28/2/765]
- 70. Benchtop versus high field NMR: Comparable performance ... [https://www.sciencedirect.com/science/article/abs/pii/S073170852200070X]
- 71. Benchtop qNMR evaluation [https://magritek.com/2016/09/18/benchtop-qnmr-evaluation/]
- 72. Comparison of HPLC and UV Spectrophotometric Methods ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8653657/]
- 73. : A Tool for Regulatory Flexibility and... Analytical Quality by Design [https://pmc.ncbi.nlm.nih.gov/articles/PMC4332986/]
- 74. Space of Design in the... - SEQENS Robust Analytical Methods [https://www.seqens.com/knowledge-center/design-space-of-robust-analytical-methods-in-the-pharmaceutical-environment/]
- 75. Recent HPLC-UV Approaches for Cannabinoid Analysis [https://www.mdpi.com/1424-8247/18/6/786]
- 76. A sustainable multi-task HPLC–UV method for ... [https://www.nature.com/articles/s41598-025-07502-8]
- 77. ICH Q2(R2) Validation of analytical procedures [https://www.ema.europa.eu/en/ich-q2r2-validation-analytical-procedures-scientific-guideline]
- 78. Key ICH Method Validation Parameters to Know [https://altabrisagroup.com/ich-method-validation-parameters/]
- 79. Analytical Methods: A Statistical Perspective on the ICH ... [https://www.biopharminternational.com/view/analytical-methods-statistical-perspective-ich-q2a-and-q2b-guidelines-validation-analytical-methods]
- 80. Comparative performance of liquid chromatography and ... [https://www.sciencedirect.com/science/article/pii/S2405844024085827]
- 81. Forensic discrimination of fiber microspectrophotometry ... [https://www.sciencedirect.com/science/article/abs/pii/S0026265X24015522]
- 82. A Review: Comparison of ICH Guidelines on Analytical ... [https://www.linkedin.com/pulse/review-comparison-ich-guidelines-analytical-method-usp-shaikh-wzfjc]
- 83. Comparative study of UV spectroscopy, RP-HPLC and HPTLC ... [https://fjps.springeropen.com/articles/10.1186/s43094-024-00651-z]
- 84. Journal of Chromatography A [https://www.sciencedirect.com/science/article/abs/pii/S0021967399013278]
- 85. UV Detection for HPLC – Fundamental Principle, Practical ... [https://www.chromatographyonline.com/view/lcgc-blog-uv-detection-hplc-fundamental-principle-practical-implications]
- 86. Are HPLC-UV Methods Fit for Purpose as True Arbiters of ... [https://www.americanpharmaceuticalreview.com/Featured-Articles/163576-Are-HPLC-UV-Methods-Fit-for-Purpose-as-True-Arbiters-of-Quality-for-APIs/]
- 87. Buy Mass Spectrometers For Sale, New & Used Prices [https://www.labx.com/categories/mass-spectrometer]
- 88. HPLC/UV approach method for the first simultaneous ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10515236/]
- 89. What is HPLC-UV? Competitors, Complementary Techs & ... [https://sumble.com/tech/hplc-uv]
- 90. Ultraviolet Detectors: Perspectives, Principles, and Practices [https://www.chromatographyonline.com/view/ultraviolet-detectors-perspectives-principles-and-practices]
- 91. HPLC, a modular technique that complements NMR [https://specificpolymers.com/hplc-a-modular-technique-that-complements-nmr/]
- 92. Metrological Evaluation of Metopimazine HPLC Assay [https://pmc.ncbi.nlm.nih.gov/articles/PMC12567434/]
- 93. Method validation and measurement uncertainty estimation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12425206/]
- 94. Comparison of ultraviolet and refractive index detections in ... [https://www.sciencedirect.com/science/article/abs/pii/S030881462101520X]
- 95. Chemometrically assisted optimization and validation of the ... [https://bmcchem.biomedcentral.com/articles/10.1186/s13065-025-01509-y]