Spectroscopy Data Analysis and Software Tools: A 2025 Guide for Biomedical Research and Drug Development
This article provides a comprehensive guide to the rapidly evolving landscape of spectroscopy data analysis, tailored for researchers, scientists, and drug development professionals.
Spectroscopy Data Analysis and Software Tools: A 2025 Guide for Biomedical Research and Drug Development
Abstract
This article provides a comprehensive guide to the rapidly evolving landscape of spectroscopy data analysis, tailored for researchers, scientists, and drug development professionals. It covers foundational principles and explores the latest software tools, including those enhanced with AI and machine learning. The scope includes practical methodologies for pharmaceutical applications, proven troubleshooting techniques for common instrumentation issues, and a critical framework for method validation and comparative analysis of techniques like ED-XRF and WD-XRF. By synthesizing current market trends and recent technological advancements, this guide aims to empower professionals to enhance data accuracy, accelerate research workflows, and meet stringent regulatory standards in biomedical and clinical research.
Mastering the Fundamentals: Core Concepts and Emerging Trends in Spectroscopy Software
Spectroscopy software has transitioned from a specialized tool for operating instruments to a critical platform for data intelligence and workflow automation. In modern laboratories, this software serves as the central nervous system, integrating with spectrometers to enable precise collection, analysis, and interpretation of spectral data across pharmaceutical development, food safety testing, and environmental monitoring [1]. The global market, valued between $1.1 billion and $1.49 billion in 2024 and 2025, reflects this growing importance, with projections indicating a rise to $2.33 billion to $2.5 billion by 2029-2034 [2] [1] [3]. This growth, driven by technological innovation and stringent regulatory demands, has made understanding the software landscape essential for researchers and drug development professionals aiming to maintain analytical excellence and competitive advantage.
Current Market Size and Future Projections
The spectroscopy software market is experiencing robust global growth, fueled by advancements in artificial intelligence, cloud computing, and increasing application across regulated industries. The following table summarizes the key market size figures and growth projections from recent industry analyses:
| Market Size & Growth Metric | 2024 Value | 2025 Value | 2032/2034 Value | Compound Annual Growth Rate (CAGR) |
|---|---|---|---|---|
| Business Research Company [2] | $1.33 billion | $1.49 billion | $2.33 billion (2029) | 12.1% (2024-2025), 11.9% (2025-2029) |
| 360iResearch [4] | $250.88 million | $277.44 million | $610.20 million (2032) | 11.75% (2024-2032) |
| Global Market Insights [1] [3] | $1.1 billion | - | $2.5 billion (2034) | 9.1% (2025-2034) |
| Marketsizeandtrends [5] | $1.2 billion | - | $2.30 billion (2033) | 7.5% (2026-2033) |
Note: Discrepancies in absolute values arise from different research methodologies and market definitions (e.g., inclusion or exclusion of related services and hardware). However, all sources consistently indicate strong, positive growth.
Market Growth Drivers and Trends
Several interconnected factors are propelling the expansion and transformation of the spectroscopy software market:
Pharmaceutical Industry Demand: The pharmaceutical sector accounted for over 28.9% of the market share in 2024 [1] [3]. The need for high-throughput screening in drug discovery, rigorous quality control, and compliance with regulatory standards is a primary driver. The FDA's approval of 55 new drugs in 2023 exemplifies the industry's pace, which relies heavily on advanced analytical tools [1].
Stringent Regulatory and Safety Requirements: Increasing global emphasis on food safety and environmental monitoring is boosting software adoption. For instance, food and beverage recalls in the U.S. rose by 8% in 2023, highlighting the need for robust contaminant detection and quality verification tools [2].
Technology Integration: The integration of Artificial Intelligence (AI) and Machine Learning (ML) is revolutionizing data analysis by enabling advanced pattern recognition, predictive analytics, and automated anomaly detection [2] [6] [1]. Furthermore, the shift toward cloud-based and hybrid deployment models offers scalability, remote access, and enhanced collaboration for geographically dispersed teams [2] [6] [4].
Rise of Portable and Connected Systems: The market is seeing growing demand for software compatible with portable and handheld spectrometers, enabling on-site analysis in fields like agriculture, forensics, and environmental monitoring [6] [7] [1]. The integration with Laboratory Information Management Systems (LIMS) and other lab platforms is also creating more efficient, connected workflows [2] [6].
Technical Support Center: Troubleshooting and FAQs
This section addresses common technical challenges researchers face when using spectroscopy software, providing practical guidance for resolving data analysis and operational issues.
Frequently Asked Questions (FAQs)
Q1: What are the primary considerations when choosing between cloud-based and on-premise spectroscopy software?
The choice depends on your data security, compliance, and collaboration needs. On-premise solutions, which dominated the market in 2024 with USD 549.5 million in revenue, offer direct control over sensitive data, which is crucial for meeting strict regulatory requirements in pharmaceuticals and healthcare [1] [3]. They also allow for deep customization and integration with existing lab systems. Cloud-based solutions provide superior scalability, remote access, and easier collaboration, and they reduce upfront capital expenditure. They are ideal for distributed teams and labs that need to process large, variable datasets flexibly [6] [4] [1].
Q2: How is AI transforming the analysis of spectroscopic data?
AI, particularly machine learning, is revolutionizing spectroscopy software by automating complex analytical tasks [6]. Key transformations include:
- Automated Pattern Recognition and Anomaly Detection: ML algorithms can rapidly identify spectral features and correlate them with specific compounds or materials, significantly reducing analysis time and increasing accuracy [6].
- Enhanced Data Processing: Deep learning models are employed for advanced spectral deconvolution, noise reduction, and baseline correction, resulting in cleaner and more reliable data [6].
- Predictive Modeling and Insights: AI-driven software can provide actionable recommendations for quality control and R&D by interpreting complex datasets, thereby supporting better decision-making [6] [1].
Q3: Our lab is implementing new spectroscopy software. What are the key steps for successful validation, particularly under GAMP 5?
For labs operating under GxP, validating software is critical. A risk-based approach, as outlined in GAMP 5, is the industry standard [8]. The process should be integrated into your project lifecycle, from planning to decommissioning. The diagram below outlines the core logical workflow for a GAMP 5 compliant validation process.
Q4: A common issue we face is poor signal-to-noise ratio in our spectral data, especially with low-concentration samples. What are the standard troubleshooting steps?
Poor signal-to-noise ratio is a frequent challenge. Follow this systematic troubleshooting workflow to identify and resolve the issue.
Q5: We are experiencing integration failures between our spectroscopy software and the Laboratory Information Management System (LIMS). What is the recommended protocol to resolve this?
Integration issues between software systems are common. The following protocol provides a detailed methodology for diagnosing and resolving these problems.
Experimental Protocol: Diagnosing Software-LIMS Integration Failures
Objective: To systematically identify and resolve communication and data transfer failures between spectroscopy software and a LIMS.
Materials and Reagents:
- Primary Systems: Spectroscopy software (e.g., Waters Connect, Genie 4.0), LIMS (e.g., LabVantage, LabWare) [2] [1] [8].
- Testing Tools: Network diagnostic tools (e.g., ping, telnet), API testing platform (e.g., Postman).
- Documentation: System configuration files, API documentation, and data mapping specifications.
Methodology:
- Verification of Network Connectivity:
- Using a command-line interface, ping the LIMS server's IP address to confirm basic network reachability.
- Use the
telnet [LIMS_Server_IP] [Port]command to verify that the specific port required for communication is open and accessible. A failed connection at this stage indicates a network firewall or configuration issue.
Authentication and Credential Validation:
- Manually test the authentication process using an API tool like Postman. Send a login request to the LIMS API endpoint with the credentials used by the spectroscopy software.
- Confirm the credentials have not expired and have the necessary permissions for data writing and reading. Note any error codes related to "unauthorized" or "forbidden" access.
Data Format and Payload Inspection:
- Capture a sample of the data payload being sent from the spectroscopy software. Compare the structure (e.g., JSON, XML schema) and field names directly against the expected format defined in the LIMS API documentation.
- Pay specific attention to data types (e.g., text vs. numeric), mandatory fields, and character limits. Mismatches here are a common source of "silent" failures where the connection is successful but data is rejected.
Log File Analysis:
- Examine the application log files of both the spectroscopy software and the LIMS. Filter for errors, warnings, or failed transactions that occurred at the time of the integration attempt.
- Cross-reference timestamps to correlate events between the two systems. The logs often provide specific error messages that are not displayed in the user interface.
Expected Outcome: By following this protocol, the root cause of the integration failure will be identified, typically falling into one of the categories above. The resolution may involve network reconfiguration, credential updates, data mapping adjustments, or software patching.
Troubleshooting Note: If the issue persists after these steps, contact the technical support teams for both the spectroscopy software and LIMS vendors, providing them with the detailed findings from this diagnostic protocol.
The Scientist's Toolkit: Essential Research Reagent Solutions
The following table details key reagents and materials frequently used in spectroscopic experiments, particularly in pharmaceutical applications.
| Item Name | Function / Role in Experiment |
|---|---|
| Ultrapure Water | Used for sample preparation, dilution, and as a blank solvent; essential for achieving low background noise in UV-Vis and FT-IR spectroscopy [7]. |
| Deuterated Solvents (e.g., D₂O, CDCl₃) | Required for Nuclear Magnetic Resonance (NMR) spectroscopy to provide a non-interfering signal lock and allow for accurate solvent suppression [1]. |
| PCR Primers & Probes | Designed using specialized software (e.g., Primer3) for specific DNA amplification in genetics and molecular biology research, with subsequent analysis often verified by spectroscopic methods [9]. |
| Monoclonal Antibodies | Key analytes in biopharmaceutical characterization; analyzed using specialized fluorescence techniques like A-TEEM for stability, aggregation, and identity testing [7]. |
| Certified Reference Materials | Provide known spectral signatures for instrument calibration, method validation, and ensuring quantitative accuracy across all spectroscopic techniques [1]. |
Troubleshooting Guides
Common FT-IR Spectroscopy Problems and Solutions
Modern spectroscopy software integrates advanced capabilities for data collection, analysis, and interpretation, but users may still encounter technical issues. The following table outlines common problems in FT-IR spectroscopy and their recommended solutions [10].
| Problem | Symptoms | Likely Cause | Solution |
|---|---|---|---|
| Instrument Vibrations | Noisy spectra; strange, unexplained peaks | Physical disturbances from nearby equipment or lab activity | Relocate spectrometer to a vibration-free surface; isolate from pumps and heavy traffic [10]. |
| Dirty ATR Crystal | Negative absorbance peaks; distorted baselines | Contaminated or dirty crystal surface | Clean the ATR crystal thoroughly with appropriate solvent and acquire a fresh background scan [10]. |
| Incorrect Data Processing | Distorted spectral output in diffuse reflection | Data processed in absorbance units instead of Kubelka-Munk | Reprocess the data, converting to Kubelka-Munk units for a more accurate analytical representation [10]. |
| Surface vs. Bulk Effects | Inconsistent results from the same material sample | Surface chemistry (e.g., oxidation) differs from bulk material | Collect spectra from both the material's surface and a freshly cut interior section for comparison [10]. |
Software and Data Integrity FAQs
Q: Our laboratory must adhere to strict data security and compliance protocols. What software deployment option is most suitable? [1] A: The on-premises deployment model is often preferred in regulated environments like pharmaceuticals and healthcare. It provides organizations with direct control over sensitive spectral data, helps meet specific regulatory requirements (e.g., FDA compliance), and allows for deeper customization and integration with existing laboratory systems [1].
Q: How can our team quickly interrogate data to plot chromatograms, perform library searches, or annotate spectra without launching a full quantitative analysis? [11] A: Many software suites, such as Thermo Scientific Xcalibur, include built-in applications for ad-hoc data review. The FreeStyle application, for example, allows users to qualitatively interrogate data by displaying chromatograms and spectra, integrating peaks, searching mass spectral libraries, and annotating plots with text and graphics [11].
Q: What are the key technological trends making spectroscopy software more powerful and accessible? [1] A: The market is rapidly evolving with several key trends:
- Integration of AI and ML: Enhances data processing speed, pattern recognition, and predictive analytics.
- Cloud-Based Solutions: Enables remote access, facilitates collaboration among geographically dispersed teams, and offers scalable computing resources.
- Intuitive User Interfaces: Development of user-friendly dashboards, automated workflows, and customizable reporting makes the software accessible to non-specialists.
- Modular Software Design: Allows users to select and pay for only the features they need, providing flexibility for evolving research requirements [1].
Q: Where can I find resources for instrument maintenance, operation, and software support? [12] A: Most instrument manufacturers provide comprehensive technical support centers. These typically offer:
- Expert Guidance: Remote or on-site help with installation, calibration, and advanced troubleshooting.
- Comprehensive Training: Sessions on basic operation and advanced software functionalities.
- Extensive Online Resources: Access to user manuals, FAQs, troubleshooting guides, and instructional videos [12]. For specific platforms, such as Thermo Fisher's Real-Time PCR systems, dedicated support centers provide technical notes, FAQs, and workflow walkthroughs [13].
Workflow Visualization
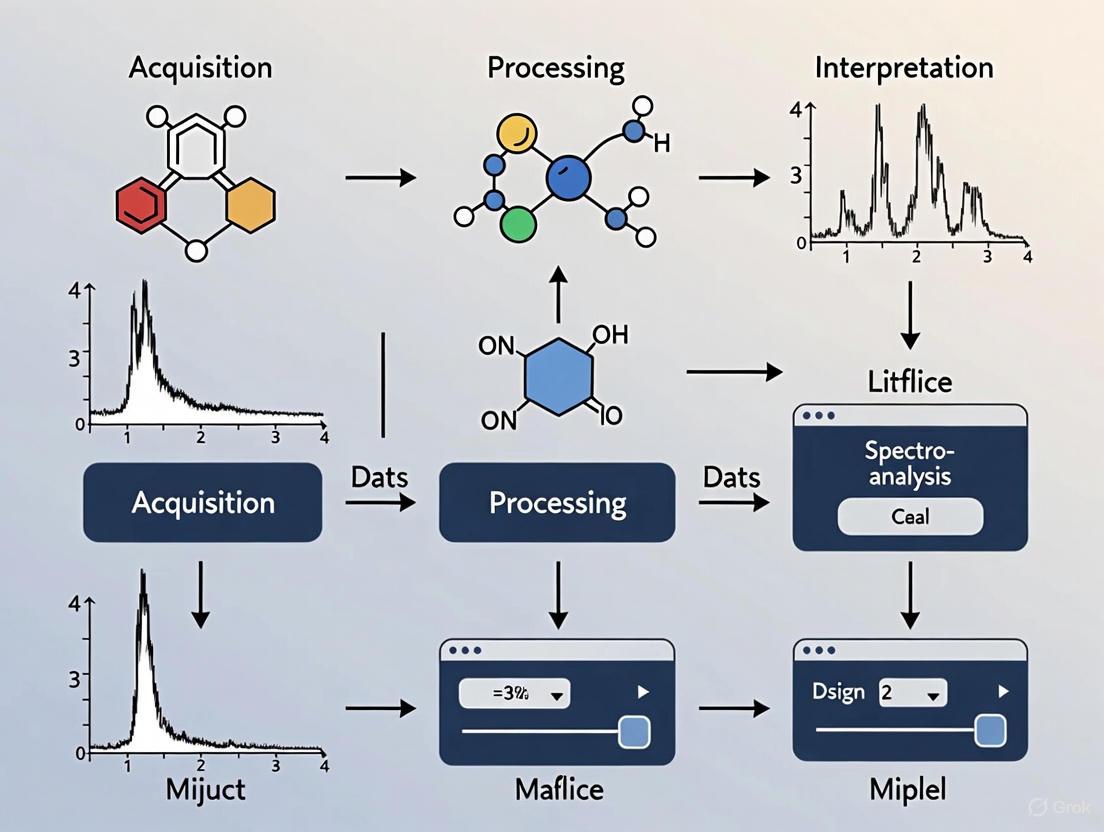
Spectroscopy Data Analysis Workflow
The following diagram illustrates the core logical workflow of spectroscopy software, from data acquisition to final reporting, highlighting the key functions at each stage.
The Scientist's Toolkit: Essential Software Solutions
The following table details key software solutions and their primary functions in the spectroscopy workflow, crucial for ensuring data integrity and analytical efficiency [1] [11] [14].
| Software/Tool | Primary Function | Key Application in Research |
|---|---|---|
| Xcalibur Software [11] | Data acquisition, control, and interrogation for LC-MS systems. | Provides a centralized platform for method setup, data review, and integration with cloud-based tools for collaborative analysis. |
| SynerJY Software [14] | Integrated data acquisition and analysis for spectroscopic systems. | Offers intuitive control of spectrometers and detectors, with advanced data processing and presentation tools like 3-D plots and contour maps. |
| LabSpec 6 Software [14] | Dedicated data acquisition and analysis suite for Raman spectroscopy. | Delivers powerful, specialized capabilities for Raman analysis, including control of modular Raman systems. |
| Cary WinUV Color [15] | Color measurement and quality control software for UV-vis spectroscopy. | Automates generation of QA/QC reports using international color coordinate systems (e.g., chromaticity, CIELAB) for industries where color consistency is critical. |
| AI/ML Enhanced Platforms [1] | Advanced data analysis and pattern detection. | Improves speed and precision in processing spectral data, enabling predictive analytics and high-throughput screening in drug discovery. |
Technical Support Center
Frequently Asked Questions (FAQs)
Q1: What is the fundamental difference between traditional chemometrics and new AI methods for spectral analysis?
Traditional chemometrics relies on linear models like Principal Component Analysis (PCA) and Partial Least Squares (PLS) regression, which are vital for transforming multivariate datasets into actionable insights [16]. In contrast, modern Artificial Intelligence (AI) and Machine Learning (ML) frameworks automate feature extraction, handle nonlinear calibration, and facilitate data fusion. Key AI subfields include Machine Learning (ML), which develops models that learn from data without explicit programming, and Deep Learning (DL), which uses multi-layered neural networks for hierarchical feature extraction [16]. This enables AI to process unstructured data like hyperspectral images and high-throughput sensor arrays more effectively.
Q2: How can I quantify the uncertainty of predictions made by my machine learning model on spectroscopic data?
You can implement Quantile Regression Forest (QRF), a machine learning technique based on Random Forest. Unlike standard models that provide only a single prediction, QRF retains the distribution of responses within its decision trees. This allows it to calculate prediction intervals and provide a sample-specific uncertainty estimate alongside each prediction [17]. For example, values near the detection limit will naturally produce larger prediction intervals, clearly communicating greater uncertainty to the user [17].
Q3: Our team struggles with interpreting "black box" AI models. What are some strategies for improving model interpretability?
Interpretability is a common challenge. You can employ Explainable AI (XAI) frameworks to identify informative wavelength regions and preserve chemical insight. Techniques include:
- SHAP (SHapley Additive exPlanations): Explains the output of any ML model by quantifying the contribution of each feature.
- Grad-CAM (Gradient-weighted Class Activation Mapping): Highlights important regions in an input, often used with convolutional neural networks.
- Spectral Sensitivity Maps: Visualize which parts of the spectrum most influence the model's decision [16]. Using these tools helps reconcile powerful AI predictions with fundamental chemical principles.
Q4: What are the best practices for visualizing magnetic resonance spectroscopy (MRS) data to ensure study validity?
A survey of the MRS literature revealed generally poor visualization standards [18]. To ensure robustness and interpretability:
- Voxel Location: Present the voxel location from all participants, not just a single example. Participant group membership (e.g., patients and controls) should be clearly indicated [18].
- Spectral Quality: Provide a visual representation of the MRS spectra from all participants, not just an average or a single spectrum. This allows readers to directly judge data quality and identify artefacts [18]. Presenting this information is essential for judging the validity and replicability of the experiment.
Q5: How can we use AI to analyze hyperspectral imaging (HSI) data cubes for pharmaceutical quality control?
Hyperspectral data cubes integrate spatial and chemical information [19]. The analysis workflow typically involves:
- Data Acquisition: Using HSI to gather spatial and spectroscopic data into a cube where the x- and y-axes are spatial, and the z-axis is spectral [19].
- Data Correction: Processing data to correct for baseline drifts, spectral overlap, and multiplicative scattering [20].
- Region of Interest (ROI) Identification: Applying unsupervised ML models like PCA to determine the ROI that best represents your analyte of interest [20].
- Classification/Quantification: Using supervised models like K-Nearest Neighbor (KNN) for classification or PLS for quantitative modeling based on the identified ROI [20].
Troubleshooting Guides
Issue 1: Poor Generalization and Overfitting in Supervised ML Models
- Problem: Your model performs well on training data but poorly on new, unseen spectral data.
- Solution:
- Increase and Diversify Training Data: Ensure your training set is sufficiently large and comprehensively covers the chemical space of interest. The application of ML to experimental data is often limited by the amount of consistent data that can be produced [21].
- Apply Regularization: Use techniques that add a penalty to complex solutions in your model to prevent it from fitting to noise in the training data [21].
- Use Ensemble Methods: Algorithms like Random Forest (RF) build many decision trees and aggregate their results, offering strong generalization capability and reduced overfitting [16].
- Validate Rigorously: Always use a hold-out test set or cross-validation to assess real-world performance.
Issue 2: Handling Nonlinear Relationships in Spectral Data
- Problem: Traditional linear models (PLS, PCR) fail to capture complex, nonlinear relationships in your dataset.
- Solution:
- Explore Nonlinear Algorithms: Implement models designed for nonlinearity, such as:
- Support Vector Machines (SVM) with nonlinear kernels (e.g., Radial Basis Function) [16].
- Artificial Neural Networks (ANNs) and Deep Neural Networks (DNNs), which can approximate complex calibration functions [16].
- Extreme Gradient Boosting (XGBoost), which sequentially corrects residual errors and excels at complex, nonlinear relationships [16].
- Preprocess Data: Apply scatter correction or normalization to mitigate physical nonlinearities like light scattering effects.
- Explore Nonlinear Algorithms: Implement models designed for nonlinearity, such as:
Issue 3: "Circumstantial" or Spurious Correlations in Chemometric Models
- Problem: The model shows a strong statistical correlation between a spectral feature and a sample property, but the correlation is not grounded in sound chemistry (e.g., predicting sulfur in gasoline with molecular spectroscopy) [22].
- Solution:
- Apply "Chemical Thinking": Reason whether the component responsible for the property can genuinely express itself spectrally in the technique you are using. Molecular spectroscopy measures chemical bonds, so it may be unsuitable for quantifying elements regardless of their molecular form [22].
- Investigate Underlying Factors: Correlations can be rooted in the interconnected nature of process data. A change in one compound might be artificially correlated with another due to the process itself, not their spectral properties [22].
- Validate with First Principles: Ensure your model has a plausible basis in chemistry and physics, not just statistics.
Essential Research Reagent Solutions
The following table details key software tools and algorithms used in modern AI-driven spectroscopic analysis.
| Research Reagent / Tool | Type | Primary Function in Analysis |
|---|---|---|
| Partial Least Squares (PLS) [20] [16] | Algorithm | A foundational supervised method for linear regression and quantitative analysis, finding latent variables that relate spectral signals to response matrices. |
| Principal Component Analysis (PCA) [20] [16] | Algorithm | An unsupervised technique for exploratory data analysis, dimensionality reduction, and clustering; essential for identifying patterns and outliers in hyperspectral data. |
| Random Forest (RF) [16] [17] | Algorithm | An ensemble learning method used for classification and regression that offers strong generalization and robustness against spectral noise and collinearity. |
| Quantile Regression Forest (QRF) [17] | Algorithm | An extension of Random Forest that provides both accurate predictions and sample-specific uncertainty estimates, crucial for reliable analytical reporting. |
| Convolutional Neural Network (CNN) [16] | Algorithm | A deep learning architecture ideal for automatically extracting hierarchical features from raw spectral data or hyperspectral images. |
| Hyperspectral Imaging (HSI) [20] [19] | Technique & Data | A measurement method that simultaneously acquires spatial and spectroscopic data, creating a data cube for detailed material characterization and mapping. |
| Explainable AI (XAI) [16] | Framework | A set of tools (e.g., SHAP, Grad-CAM) used to interpret complex AI models, identifying which spectral features drive predictions to maintain chemical insight. |
Experimental Protocol: Developing a QRF Model for Quantitative Spectral Analysis
This protocol outlines the methodology for applying a Quantile Regression Forest to predict sample properties and estimate prediction uncertainty from infrared spectroscopic data, as demonstrated in soil and agricultural analysis [17].
1. Sample Preparation and Spectral Acquisition
- Collect a representative set of samples (e.g., soil, agricultural produce, pharmaceutical blends).
- For each sample, acquire the infrared spectrum (e.g., NIR, FTIR) using a calibrated spectrometer. Ensure consistent measurement conditions (e.g., pathlength, temperature) across all samples.
- In parallel, use a primary test method (PTM) to obtain reference values for the property of interest (e.g., cation exchange capacity, dry matter content) for each sample [22].
2. Dataset Construction and Preprocessing
- Construct a dataset where each entry is a paired spectrum and its corresponding reference value.
- Randomly split the dataset into a calibration (training) set (typically 70-80% of samples) and a validation (test) set (the remaining 20-30%).
- Apply necessary spectral pre-processing to the calibration set, such as smoothing, baseline correction, or standard normal variate (SNV) transformation. Critical: The parameters for these pre-processing steps must be derived from the calibration set only and later applied to the validation set to avoid data leakage.
3. Model Training and Calibration
- Train the Quantile Regression Forest model on the pre-processed calibration set.
- The model will learn the relationship between the spectral features (X) and the reference values (Y) from the PTM.
- Unlike standard Random Forest, QRF retains the full distribution of the response variables in the leaves of its trees, which is essential for uncertainty estimation [17].
4. Model Validation and Uncertainty Estimation
- Use the trained QRF model to predict the property values for the hold-out validation set.
- For each prediction, the model will also output a prediction interval (e.g., a 90% interval). This interval provides a range within which the true value is likely to fall, offering a sample-specific measure of uncertainty [17].
- Assess the model's performance by comparing the predicted values to the known reference values from the PTM for the validation set. Calculate metrics like Root Mean Square Error (RMSE) and R².
5. Interpretation and Operational Use
- Examine the validation results. Note that some predictions, especially for extreme values or samples near the model's detection limit, will have wider prediction intervals, indicating higher uncertainty [17].
- The model is ready for operational use once it demonstrates satisfactory predictive accuracy and reliable uncertainty quantification on the validation set.
The workflow for this protocol is summarized in the following diagram:
For researchers, scientists, and drug development professionals, the choice of software deployment model is a critical strategic decision that directly impacts the efficiency, security, and scalability of spectroscopy data analysis. The core of this decision often involves a fundamental trade-off: the extensive control and data security offered by on-premises solutions versus the unparalleled scalability and collaborative flexibility of cloud platforms. In the context of spectroscopy, where data integrity and regulatory compliance are paramount, understanding this balance is essential. This guide provides a technical support framework to help scientific teams navigate this complex landscape, troubleshoot common issues, and implement best practices tailored to analytical research environments.
Core Concepts and Definitions
- On-Premises Deployment: The software and all associated data are hosted on infrastructure located within the organization's own facilities (e.g., a local server room or data center). The organization is entirely responsible for all maintenance, security, updates, and physical access control [23] [24].
- Cloud Deployment: The software is hosted on the service provider's remote servers and accessed over the internet. The provider manages the infrastructure, and resources are typically delivered via a subscription-based, pay-as-you-go model [23] [25]. Common models include Software as a Service (SaaS), which is most prevalent for end-user applications.
- Spectroscopy Data Analysis: The process of using specialized software tools to collect, process, and interpret spectral data from instruments like spectrometers. This software is crucial for determining material composition and is widely used in drug discovery, quality assurance, and process control [1].
Comparative Analysis: On-Premises vs. Cloud
The following tables summarize the key differences between the two deployment models, with a focus on aspects critical to research and development environments.
Table 1: Strategic Comparison of Deployment Models
| Parameter | On-Premises Security | Cloud Security |
|---|---|---|
| Data Location & Control | Data resides on internal servers, providing complete physical and logical control over data and encryption keys [23]. | Data is stored in the vendor's remote data centers; users have less direct control, which is managed by a third-party provider [23]. |
| Customization | Solutions can be highly customized to specific research workflows and integrated with existing laboratory systems [23]. | Offers limited customization, typically confined to the features and configurations provided by the vendor [23]. |
| Compliance | Often preferred for heavily regulated industries (e.g., pharma, healthcare) as it simplifies meeting strict data sovereignty and audit requirements like HIPAA and GDPR [23] [1]. | Providers offer compliance certifications, but the shared responsibility model requires users to ensure their configuration and use meet regulatory standards [23] [26]. |
| Software Updates | The internal IT team has full control over the timing and implementation of upgrades and patches [24]. | The provider manages all software updates and patches automatically, ensuring access to the latest features without manual intervention [25]. |
Table 2: Quantitative Market Data for Spectroscopy Software (2024)
| Aspect | On-Premises Deployment | Cloud Deployment & Market Trends |
|---|---|---|
| Market Size (2024) | USD 549.5 Million [1] | Part of a global market valued at USD 1.1 Billion [1] |
| Growth Driver | Data security and compliance capabilities, particularly in pharmaceuticals and healthcare [1]. | Incorporation of AI and ML for data analysis, and the rise of remotely accessible solutions for collaboration [1]. |
| Key Advantage | Direct control over sensitive information and faster data processing for time-sensitive applications [1]. | Scalability of storage and computing resources to handle increasing volumes of spectral data [1]. |
Decision Framework and Workflow
Choosing the right deployment model requires a systematic assessment of your project's specific needs. The following diagram outlines the key decision-making workflow.
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Software and Hardware Components for Spectroscopy
| Item | Function in Spectroscopy |
|---|---|
| FT-IR Spectrometer | A core instrument for collecting molecular fingerprint data; new platforms (e.g., Bruker Vertex NEO) enhance performance by minimizing atmospheric interference [7]. |
| Spectroscopy Software Suite | Specialized tools (from vendors like Thermo Fisher, Agilent) for instrument control, spectral data acquisition, processing, and interpretation [1]. |
| Laboratory Information Management System (LIMS) | Software that tracks samples and associated data, ensuring workflow integrity and regulatory compliance [27]. |
| Quantum Cascade Laser (QCL) Microscope | An advanced tool (e.g., Bruker LUMOS II) for high-resolution infrared imaging of micro-samples, crucial for pharmaceutical analysis [7]. |
| Cloud-Native Data Analysis Platform | A platform that provides centralized, remotely accessible storage and built-in AI/ML tools for processing high volumes of spectral data [1]. |
Troubleshooting Guides and FAQs
Frequently Asked Questions
Q1: Our team is geographically dispersed. How can we securely collaborate on the same spectral data sets in real-time?
A1: Cloud platforms are inherently designed for this challenge. They provide a centralized repository for spectral data that authorized users can access from any location with an internet connection. This enables real-time collaboration on data analysis and shared projects. To ensure security, implement Role-Based Access Control (RBAC) to define precisely which data and functions each researcher can access, adhering to the principle of least privilege [28] [29]. All data in transit and at rest should be protected with enterprise-grade encryption [25].
Q2: We have legacy instrumentation and custom analysis scripts. Which deployment model offers better integration?
A2: On-premises solutions typically excel here. They offer a higher degree of customization and direct system access, making it easier to integrate with specialized legacy equipment and execute custom scripts or workflows that may not be compatible with standardized cloud environments [23] [24]. The controlled local network can also provide the low-latency connection sometimes required for direct instrument control [30].
Q3: We are experiencing slow performance when analyzing large hyperspectral imaging datasets. How can we improve processing speed?
A3:
- If using an on-premises system: Investigate upgrading your local hardware, specifically the CPU, GPU, and RAM, as these directly impact processing speed for large datasets [30]. Ensure your IT team performs regular maintenance and that no other network-intensive processes are consuming resources.
- If using a cloud platform: Leverage its scalability. Most cloud services allow you to temporarily provision more powerful computing resources (e.g., high-memory or GPU-optimized instances) specifically for the duration of the intensive analysis task, scaling back down afterward to manage costs [28] [26].
Q4: How do we ensure our spectroscopy data management is compliant with regulations like FDA 21 CFR Part 11?
A4: Compliance is a shared responsibility. For on-premises deployments, the organization has full control to implement the required technical controls (e.g., audit trails, electronic signatures, access security) and can more easily maintain detailed records for audits, as all data resides internally [23] [1]. When using a cloud service, you must select a provider that explicitly offers compliance certifications relevant to your industry (e.g., HIPAA, GDPR). Crucially, you are responsible for configuring the cloud application and user access in a way that maintains compliance with these regulations [26].
Q5: Our internet connection is unstable. What is the impact on cloud-based spectroscopy software, and what are the alternatives?
A5: Unstable internet will render cloud software inaccessible during outages and can cause severe lag or data transfer failures, disrupting research activities [24] [26]. In such scenarios, the most robust alternative is a hybrid approach. In this model, primary data analysis workflows run on an on-premises server to ensure uninterrupted operation. The cloud can then be used for specific tasks that benefit from its power, such as long-term data archiving, running occasional large-scale AI-driven analyses, or sharing finalized results with external partners, which can be scheduled for periods of stable connectivity [30].
The field of spectroscopy is undergoing a significant transformation, characterized by two powerful, interconnected trends: the rapid adoption of portable and handheld spectrometers and the development of increasingly intelligent, user-friendly software dashboards. This shift is moving analytical power from centralized laboratories directly into the field and onto the production floor, enabling real-time, data-driven decision-making. For researchers and drug development professionals, this evolution is not merely about convenience; it enhances high-throughput screening, facilitates on-site quality assurance, and accelerates research and development cycles. The global spectroscopy software market, valued at approximately USD 1.1 billion in 2024 and projected to grow at a compound annual growth rate (CAGR) of 9.1% to reach USD 2.5 billion by 2034, underscores the critical role of advanced data analysis in this ecosystem [1].
This technical support center is designed to help scientists navigate this new paradigm. It provides immediate troubleshooting for common hardware and software issues and serves as a knowledge base framed within the context of broader research on spectroscopy data analysis and software tools. The following sections offer detailed guides, protocols, and visual aids to ensure you can leverage these emerging technologies effectively and confidently.
Technical Support & Troubleshooting Guides
Frequently Asked Questions (FAQs)
Q1: What are the key advantages of modern spectroscopy software dashboards? Modern software platforms, such as OMNIC Paradigm, are designed to streamline analysis through user-friendly dashboard screens that provide quick access to recent work and data processing tools. Key features include a visual, drag-and-drop workflow creator, one-click library creation, and pre-defined reporting templates, all aimed at simplifying data acquisition, processing, and interpretation [31].
Q2: My portable spectrometer is producing a weak or inconsistent signal. What should I check? A weak signal is a common issue. First, verify that the laser power (for Raman) or light source is set to an appropriate level for your sample. Second, inspect and clean the optics and sampling window, as dust or fingerprints can scatter light. Finally, ensure the laser is properly focused and that the optical components are aligned according to the manufacturer's manual [32].
Q3: How can cloud-based spectroscopy software enhance collaboration? Cloud-enabled software like OMNIC Anywhere allows researchers to view, analyze, and share spectral files from any device or location. It provides a centralized platform (e.g., with free starting storage of 10 GB) where team members can comment on results, which is invaluable for geographically dispersed teams in drug development and research [31].
Q4: My spectra show excessive noise or broad, fluorescent backgrounds. What can I do? This is often due to sample fluorescence or ambient light interference. To mitigate this, you can optimize the integration time to improve the signal-to-noise ratio, use the instrument's background subtraction feature, and perform measurements in a darkened environment to reduce ambient light [32]. For Raman systems, the choice of excitation wavelength may also be a factor [33].
Q5: What routine maintenance is critical for portable spectrometers? Regular maintenance is essential for consistent performance. Key tasks include:
- Cleaning optics and the sampling window regularly with a lint-free cloth and optical-grade solution.
- Regular calibration using certified reference materials.
- Inspecting and replacing consumables like filters and O-rings as needed.
- Storing the instrument in a clean, dry environment when not in use [32].
Troubleshooting Common Hardware Issues
Table 1: Troubleshooting Portable and Handheld Spectrometers
| Problem | Possible Explanation | Recommended Solution |
|---|---|---|
| Weak/No Signal | Laser is off; dirty optics; misalignment; computer communication error [33] [32]. | Check laser is on; clean sampling window/optics; verify focus/alignment; restart software/check USB [32]. |
| Spectral Noise/Artifacts | Fluorescence; CCD saturation; ambient light; insufficient integration time [33] [32]. | Adjust integration time; use background subtraction; perform measurement in dark; defocus beam if saturated [32]. |
| Inaccurate Calibration | Calibration drift over time; software requires update [32]. | Recalibrate with certified reference materials; update instrument firmware/software [32]. |
| Peak Locations Incorrect | System is not calibrated or requires verification [33]. | Perform system verification/calibration using a known standard (e.g., verification cap, isopropyl alcohol) [33]. |
| Software "Unable to Find Device" | Communication driver error; incorrect software settings [33]. | Shut down and restart software; check device manager for hardware; reinstall drivers if necessary [33]. |
Troubleshooting Common Software and Data Analysis Issues
Table 2: Troubleshooting Spectroscopy Software and Data
| Problem | Possible Explanation | Recommended Solution |
|---|---|---|
| Poor Model Performance | Unprocessed data with outliers; suboptimal algorithm [34]. | Preprocess data (smoothing, baseline correction); remove outliers; test multiple regression algorithms [34]. |
| Difficulty Identifying Unknowns | Sample is a mixture; not in library [31]. | Use software's multi-component search (mixture analysis)功能; leverage functional group info for classification [31]. |
| Data Collaboration Challenges | Using non-cloud, localized software [31]. | Utilize cloud-based platforms (e.g., OMNIC Anywhere) for centralized data sharing and project management [31]. |
| Complex Data Interpretation | Overlapping peaks in complex samples [32]. | Compare with spectral libraries; apply multivariate analysis (e.g., PCA, PLS) [31] [32]. |
Experimental Protocols & Methodologies
Protocol: Non-Destructive Chlorophyll Content Prediction in Leaves Using a Portable NIR System
This protocol, adapted from a 2023 study, details the use of a custom IoT-based portable NIR device for predicting chlorophyll content, demonstrating the application of portable spectroscopy in agricultural science [34].
1. Hypothesis: The spectral data collected from a portable near-infrared spectrometer can be used to build a reliable predictive model for the chlorophyll content in Hami melon leaves, providing a non-destructive alternative to traditional methods.
2. Research Reagent Solutions & Materials
Table 3: Essential Materials for Portable Leaf Analysis
| Item | Function/Description |
|---|---|
| Portable NIR Spectrometer | The core sensor (e.g., AS7341 used in the study) for collecting spectral data in the field [34]. |
| Chlorophyll Meter | A validated device (e.g., Top Cloud-agri TYS-4N) for measuring reference SPAD values [34]. |
| Leaf Fixing Plate | Ensures consistent and equidistant positioning of the leaf relative to the sensor for reproducible data [34]. |
| Cloud Server/Data Platform | For data reception, storage, and processing using services like EMQX, Node-RED, and InfluxDB [34]. |
| Lint-Free Cloths | For cleaning the spectrometer's window to prevent data drift caused by contaminants. |
3. Methodology:
- Step 1: System Setup. Deploy the cloud data server using containerization (e.g., Docker) to handle MQTT communication, data flow orchestration (Node-RED), and storage (InfluxDB). Configure the portable spectrometer with its microcontroller (e.g., ESP8266-12F) to transmit data via WiFi to this server [34].
- Step 2: Sample Preparation & Data Collection. Select 100 or more leaf samples from plants at different growth stages. For each sample, first measure the chlorophyll content using the chlorophyll meter, avoiding major veins. Immediately after, place the leaf on the fixing plate and collect the spectral data using the portable device, ensuring the hardware acquisition parameters (e.g., acquisition times) are set via the web interface [34].
- Step 3: Data Preprocessing. Apply preprocessing algorithms to the raw spectral data to reduce noise. Methods can include smoothing, baseline correction, and normalization. Use algorithms like the Isolation Forest to detect and remove spectral outliers from the dataset [34].
- Step 4: Model Building & Validation. Split the data into training and prediction sets. Test a suite of regression algorithms (e.g., Linear Regression, Decision Tree, Support Vector Regression, Random Forest) on the data to identify the best-performing model. Evaluate model performance using metrics like Root Mean Square Error (RMSE) and the Coefficient of Determination (R²) for both the training and prediction sets [34].
The workflow for this experimental protocol is summarized in the diagram below:
Protocol: On-Site Material Verification with a Handheld Raman Spectrometer
This protocol outlines a standard operating procedure for verifying materials using a handheld Raman spectrometer, a common task in pharmaceutical and forensic fields.
1. Hypothesis: A handheld Raman spectrometer can quickly and accurately identify an unknown solid material by matching its spectral fingerprint against a built-in library.
2. Methodology:
- Step 1: Instrument Preparation. Ensure the spectrometer is fully charged. Turn on the device and allow it to initialize. Perform a quick verification check using a known standard (e.g., a verification cap or isopropyl alcohol) to confirm the wavelength calibration is accurate [33].
- Step 2: Sample Presentation. Place the solid sample on a clean, flat surface. For best results, the sample should have a flat and clean surface. If the sample is in a container, ensure the container is transparent at the laser wavelength and does not produce a interfering Raman signal.
- Step 3: Data Acquisition. Firmly press the spectrometer's sampling window against the sample. Ensure a good seal to block ambient light. Trigger a measurement. The integration time may be automatically set or may need to be manually optimized to obtain a spectrum with a good signal-to-noise ratio without saturating the detector [32].
- Step 4: Data Analysis and Reporting. The instrument's software will automatically compare the acquired spectrum against its spectral library and present a list of potential matches with confidence scores. Visually inspect the match between the sample spectrum and the top library hit to confirm the identification. Generate a report using the software's built-in template, which can include the sample spectrum, the matched library spectrum, and relevant sample information.
The Scientist's Toolkit: Key Technologies Driving the Trends
Portable Spectrometer Technologies
Table 4: Comparison of Portable Spectrometer Technologies
| Technology | Typical Applications | Example Products | Key Features |
|---|---|---|---|
| Handheld NIR | Pharmaceutical QA, agriculture, chemical ID [7]. | SciAps ReveNIR, Metrohm OMNIS NIRS [35] [7]. | Non-destructive; rapid material verification; minimal sample prep [35]. |
| Handheld Raman | Hazmat response, raw material ID, forensics [7]. | Metrohm TaticID-1064ST [7]. | Library-based ID; through-container testing; 1064 nm laser reduces fluorescence [7]. |
| Handheld LIBS | Alloy analysis, geochemistry, light elements (Li, Be) [35]. | SciAps Z-Series [35]. | Fast elemental analysis; particularly effective for light elements [35]. |
| Handheld XRF | Scrap metal sorting, mining, environmental monitoring [35]. | SciAps X-Series [35]. | Lab-quality elemental results in seconds; robust field design [35]. |
| Field-Portable Spectroradiometer | Environmental monitoring, geology, remote sensing [35]. | ASD Range [35]. | Full-range UV/Vis/NIR/SWIR (350-2500 nm); high signal-to-noise ratio [35]. |
Spectroscopy Software Platforms
Table 5: Comparison of Spectroscopy Software Platforms
| Software | Deployment | Key Features | Target Audience |
|---|---|---|---|
| OMNIC Paradigm (Thermo Fisher) | On-premises/Desktop [31]. | Drag-and-drop workflows; multi-component search; quantification tools; diagnostic tools [31]. | Lab managers, industrial scientists, educators [31]. |
| OMNIC Anywhere (Thermo Fisher) | Cloud-based [31]. | Cross-platform (PC, Mac, iOS, Android); data sharing & collaboration; 10GB+ free storage [31]. | Research teams, students, collaborative projects [31]. |
| Vernier Spectral Analysis | App-based (Windows, macOS, Chromebook) [36]. | Free app; simplified Beer's law & kinetics; designed for educational use [36]. | Students, educational institutions [36]. |
| WISER (Caltech) | On-premises/Desktop [37]. | Open-source; imaging spectroscopy analysis; modular plugin API; supports GEOTIFF/PDS [37]. | Researchers (Earth & planetary science) [37]. |
The integration of hardware and software, powered by AI, is creating a seamless workflow from measurement to insight, as shown below.
Applied Workflows: Advanced Techniques for Pharmaceutical and Biomedical Analysis
Troubleshooting Guides and FAQs
This section addresses common challenges researchers face when using spectroscopic techniques in pharmaceutical development.
Frequently Asked Questions
Q1: What is the first step when I obtain an Out-of-Specification (OOS) spectroscopic result? Your initial action must be to notify your supervisor and preserve the original data. A formal laboratory investigation must be initiated immediately. The first phase is an informal assessment where the analyst and supervisor review the testing procedure, calculations, instrumentation, and the notebooks containing the OOS result. A retest should not be performed until this initial investigation is complete [38].
Q2: Can I use an outlier test to invalidate an initial OOS result in chemical assays? The use of outlier tests is highly restricted. According to FDA guidance, outlier tests are inappropriate for chemical testing results and for statistically based tests like content uniformity and dissolution. An initial OOS result cannot be invalidated solely based on a statistical outlier test [38].
Q3: How many retests are permissible for an OOS result? The court provides explicit limitations on retesting. You cannot simply conduct two retests and base a release decision on the average of three tests. The investigation must determine if the original result was due to a laboratory error. The number of retests should be specified in a pre-defined, scientifically justified procedure, not determined ad-hoc [38].
Q4: What are the key trends in spectroscopy software that can enhance our lab's capabilities? Key trends include the integration of Artificial Intelligence (AI) and Machine Learning (ML) for improved data processing and predictive analytics, a shift towards cloud-based and remotely accessible solutions for collaboration, and the development of more intuitive user interfaces and automated workflows. There is also a growing emphasis on software for portable and handheld spectrometers for on-site analysis [1].
Q5: Our team struggles with overlapping spectra in chromatography. What solutions are available? Deconvolution software is designed specifically for this challenge. Tools and tutorials, such as those offered by CHROMacademy, are available to help "demystify deconvolution" and make sense of overlapping spectral data. These software solutions use algorithms to separate co-eluting peaks for accurate identification and quantification [39].
Troubleshooting Common Spectral Data Issues
Issue: Poor Signal-to-Noise Ratio in FT-IR Analysis of Proteins
- Potential Cause: Inefficient detector or atmospheric interference (water vapor, CO₂).
- Solution: Utilize a vacuum optics system, like that found in the Bruker Vertex NEO platform, which removes atmospheric contributions. Ensure the detector is appropriate for the spectral range and that the instrument is properly purged [7].
Issue: Inconsistent Results in High-Throughput Screening with Raman
- Potential Cause: Poor calibration or plate positioning errors.
- Solution: Implement a fully automated system like the PoliSpectra Raman plate reader, which integrates liquid handling and dedicated software to ensure consistency across 96-well plates. Establish a rigorous and frequent calibration schedule [7].
Issue: Data Security Concerns with Spectral Data Management
- Potential Cause: Using cloud-based systems that may not meet corporate security policies.
- Solution: Many organizations, especially in pharmaceuticals, prefer on-premises deployment of spectroscopy software. This provides direct control over sensitive data and helps meet stringent regulatory requirements like FDA 21 CFR Part 211 [1].
Spectroscopy Software Market and Application Data
The following tables summarize quantitative data on the spectroscopy software market and its primary applications, highlighting its critical role in the pharmaceutical industry.
Table 1: Global Spectroscopy Software Market Overview [1]
| Metric | Value / Share | Timeframe / Context |
|---|---|---|
| Market Size in 2024 | USD 1.1 Billion | Base Year 2024 |
| Projected CAGR | 9.1% | Forecast Period 2025–2034 |
| Market Size in 2034 | USD 2.5 Billion | Projected Value |
| Pharmaceutical Segment Share | 28.9% | Share of market in 2024 |
| Leading Deployment Model | On-Premises (USD 549.5 Million Revenue) | 2024 Revenue |
| U.S. Market Revenue | USD 310.2 Million | 2024 Revenue |
Table 2: Key Techniques and Applications in Drug Discovery & Development [40] [7]
| Therapeutic Modality | Key Spectroscopic Techniques | Primary Application in Development |
|---|---|---|
| Small Molecule Pharmaceuticals | NMR, FT-IR, UV-Vis | Solid polymorph identification, purity analysis, chemical stability |
| Protein Biologics & Vaccines | Fluorescence (A-TEEM), Raman, QCL Microscopy | Higher Order Structure (HOS) analysis, aggregation kinetics, stability |
| mRNA & Lipid Nanoparticles (LNPs) | Small-Angle Scattering, Atomic Force Microscopy | Particle structure, component location in complex systems |
| All Modalities | LC-MS, GC-MS | Impurity identification, quantification, and fate mapping |
Experimental Protocols for Pharmaceutical Analysis
Protocol 1: Impurity Fate and Purge Analysis Using Hyphenated LC-MS and Data Visualization
This protocol is critical for ensuring drug product safety by tracking the formation and removal of process-related impurities [41].
1. Objective: To identify, quantify, and track the "fate" of synthetic impurities through various synthesis and purification steps to ensure they are purged to acceptable levels.
2. Materials and Software:
- Luminata Software (ACD/Labs) or equivalent for impurity fate mapping [41].
- Liquid Chromatograph coupled to a Mass Spectrometer (LC-MS).
- Samples from each stage of the chemical synthesis process (starting materials, intermediates, reaction mixtures, and purified active pharmaceutical ingredient (API)).
3. Methodology:
- Step 1: Sample Acquisition: Collect representative samples from every unit operation in the synthetic process.
- Step 2: LC-MS Analysis: Analyze all samples using a consistent, validated LC-MS method to separate and detect components.
- Step 3: Data Integration: Import all chromatographic and spectral data into the visualization software. Link each spectral data set to the corresponding chemical structure and process step.
- Step 4: Impurity Mapping: Use the software's interactive process map to visualize the presence and concentration of each impurity at each stage.
- Step 5: Purge Calculation: The software will quantitatively determine the purge factor for each impurity, calculating its reduction over several process steps.
4. Data Interpretation: The resulting fate map provides a visual confirmation of which impurities are effectively removed and which may require additional control strategies. This is a core component of a Quality by Design (QbD) approach to process development [41].
Protocol 2: Protein Aggregation Study Using Quantum Cascade Laser (QCL) Microscopy
This protocol uses advanced infrared microscopy to monitor protein stability, a key concern for biologic therapeutics [7].
1. Objective: To identify and characterize protein aggregates within a formulated biologic drug product to assess its stability and shelf-life.
2. Materials:
- QCL-based Infrared Microscope (e.g., Bruker LUMOS II or ProteinMentor system).
- Attenuated Total Reflection (ATR) crystal.
- Sample of the biologic product (e.g., a monoclonal antibody formulation).
3. Methodology:
- Step 1: Sample Preparation: Place a small droplet (e.g., 2-10 µL) of the protein formulation onto the ATR crystal. Allow it to air-dry to form a thin film for analysis.
- Step 2: Spectral Acquisition: Using the microscope, acquire infrared spectra in the mid-infrared range (1800-1000 cm⁻¹). For imaging, define a region of interest and collect a hyperspectral image cube using the focal plane array detector.
- Step 3: Data Analysis: Analyze the amide I band (≈1650 cm⁻¹) for shifts in shape or position, which indicate changes in secondary structure. Use statistical analysis (e.g., principal component analysis) on the hyperspectral data to identify spatial regions with different spectral signatures, corresponding to aggregates.
4. Data Interpretation: The presence of aggregates will be indicated by distinct spectral features in the amide bands. The QCL microscope's speed and sensitivity allow for the detection of even small, localized aggregates that might be missed by bulk analysis techniques.
Experimental Workflow Visualization
The following diagrams illustrate the logical workflow for key spectroscopic analyses in pharmaceutical quality control and drug development.
Diagram 1: FDA OOS Investigation Workflow
Diagram 2: Impurity Fate Mapping Workflow
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Reagents, Software, and Instrumentation for Spectroscopic Analysis
| Item / Solution | Function / Application | Example Products / Technologies |
|---|---|---|
| Ultrapure Water System | Preparation of mobile phases, buffers, and sample dilution to prevent interference. | Milli-Q SQ2 Series [7] |
| Spectrofluorometer | Protein stability analysis, vaccine characterization, and monitoring molecular interactions. | Edinburgh Instruments FS5 v2; HORIBA Veloci A-TEEM [7] |
| QCL Infrared Microscope | High-sensitivity detection of protein aggregates and chemical impurities in biopharmaceuticals. | Bruker LUMOS II; ProteinMentor system [7] |
| Data Visualization Software | Impurity fate mapping, linking chemical structures to analytical data for QbD. | ACD/Labs Luminata [41] |
| Cheminformatics Toolkit | Managing chemical libraries, virtual screening, and SAR analysis in drug discovery. | RDKit (Open-Source) [42] |
| Handheld Raman Spectrometer | On-site raw material identification and quality control in the warehouse or production line. | Metrohm TaticID-1064ST [7] |
Hybrid analytical techniques represent a powerful paradigm in modern instrumentation, combining the separation capabilities of chromatography with the identification and quantification powers of mass spectrometry (MS) and spectroscopy. These integrated systems, such as GC-MS, LC-MS, GC-IR, and LC-NMR, have revolutionized chemical analysis by enabling precise characterization of complex mixtures in fields ranging from pharmaceuticals and environmental monitoring to food safety and clinical diagnostics [43]. The core strength of these hybrid platforms lies in their synergistic operation: chromatography efficiently separates individual components in a mixture, while the coupled spectroscopic or spectrometric detector provides detailed structural information for each separated analyte [43].
The global market for spectroscopy software, valued at approximately $1.1 billion in 2024 and projected to grow at a compound annual growth rate (CAGR) of 9.1% through 2034, underscores the critical importance and expanding adoption of these technologies [1]. This growth is largely driven by technological advancements, including the integration of artificial intelligence (AI) and machine learning (ML) into spectroscopy software, which enhances data processing, pattern recognition, and predictive analytics capabilities [1]. Furthermore, the pharmaceutical industry represents a major end-user, accounting for 28.9% of the spectroscopy software market share in 2024, highlighting its essential role in drug discovery and quality control [1].
Technical Support Center: Troubleshooting Guides and FAQs
GC-MS Troubleshooting Guide
Common Problem: No Peaks or Loss of Sensitivity
- Potential Cause and Solution: Gas leaks can cause sensitivity loss and sample contamination. Systematically check for leaks at the gas supply filter, shutoff valves, EPC connections, weldment lines, and column connectors using a leak detector. Retighten connections or replace cracked components as needed [44].
- Potential Cause and Solution: A blocked injection needle or syringe can prevent the sample from reaching the detector. Flush the needle or replace it if necessary. Also, verify that the auto-sampler is functioning correctly and that the sample is properly prepared [44].
- Potential Cause and Solution: Check the column for cracks, which would prevent analytes from reaching the detector. Ensure the MS detector flame is lit (if applicable) and that all gases are flowing at the correct rates [44].
Common Problem: High System Pressure
- Potential Cause and Solution: A blockage in the chromatographic column is a frequent culprit. Attempt to backflush the column with a strong organic solvent. If pressure remains high, replace the guard column and/or the analytical column [45].
- Potential Cause and Solution: The system's in-line filter may be blocked. Replace the filter to restore normal flow and pressure [45].
LC-MS Troubleshooting Guide
Common Problem: Baseline Noise or Drift
- Potential Cause and Solution: Check for loose fittings throughout the system and tighten them gently. Also, inspect pump seals and replace them if they appear worn out [45].
- Potential Cause and Solution: Air bubbles in the mobile phase or system can cause significant noise and drift. Degas the mobile phase thoroughly and purge the LC system to remove air [45].
- Potential Cause and Solution: A contaminated or aging detector flow cell can introduce noise. Clean the flow cell with a strong organic solvent. If the problem persists, the detector lamp may be low on energy and require replacement [45].
Common Problem: Peak Tailing or Broadening
- Potential Cause and Solution: Active sites or a blockage within the chromatographic column can cause peak tailing. Reverse-flush the column with a strong solvent or replace it. Using a different stationary phase chemistry may also help [45].
- Potential Cause and Solution: Inappropriate mobile phase pH or composition can lead to poor peak shape. Prepare a fresh mobile phase with the correct pH and buffer concentration [45].
- Potential Cause and Solution: An excessively long or wide internal diameter tubing between the column and the detector can broaden peaks. Minimize this post-column volume by using shorter, narrower tubing [45].
Common Problem: Fluctuating or Unstable Pressure
- Potential Cause and Solution: Air trapped in the pump or check valve malfunction can cause pressure fluctuations. Degas all solvents thoroughly, purge the pump, and inspect or replace the check valves [45].
- Potential Cause and Solution: A partial blockage in the injector or a failing pump seal can cause pressure instability. Flush the injector and tubing. If needed, replace the pump seal [45].
FT-IR Spectroscopy Troubleshooting Guide
Common Problem: Noisy Spectra
- Potential Cause and Solution: Instrument vibration is a common source of noise. FT-IR spectrometers are highly sensitive to physical disturbances from nearby equipment like pumps or general lab activity. Ensure the instrument is placed on a stable, vibration-free surface [10].
Common Problem: Negative Absorbance Peaks
- Potential Cause and Solution: A dirty Attenuated Total Reflection (ATR) crystal is the most likely cause. Contaminants on the crystal surface can scatter light and produce anomalous peaks. Clean the crystal carefully according to the manufacturer's instructions and collect a fresh background spectrum [10].
Common Problem: Distorted or Inaccurate Spectral Features
- Potential Cause and Solution: The analyzed surface may not be representative of the bulk material (e.g., due to surface oxidation or additives). For reliable results, compare spectra from the material's surface with spectra collected from a freshly cut interior section [10].
- Potential Cause and Solution: Using incorrect data processing modes can distort spectral outputs. For example, when using diffuse reflection, data should be processed in Kubelka-Munk units rather than absorbance for an accurate representation [10].
General Mass Spectrometry Troubleshooting
Common Problem: Loss of Sensitivity or Signal
- Potential Cause and Solution: Source contamination is a frequent issue in MS. Regularly clean the ion source according to the instrument manufacturer's scheduled maintenance protocol. Using high-quality, clean solvents and samples can prevent premature contamination [44].
- Potential Cause and Solution: Incorrect tuning or calibration can lead to sensitivity loss. Perform routine mass calibration and tuning using the recommended calibration standards to ensure the instrument is operating optimally [46].
Essential Research Reagent Solutions
The following table details key consumables and reagents critical for ensuring the reliability and reproducibility of experiments using hybrid analytical techniques.
Table 1: Essential Research Reagents and Consumables
| Item | Function in Hybrid Analysis |
|---|---|
| High-Purity Chromatography Vials and Caps | Designed for precision and reliability in demanding applications, they prevent sample contamination and evaporation, ensuring accuracy and repeatability in GC and LC analyses [43]. |
| Ultrapure Water (e.g., from Milli-Q SQ2 systems) | Essential for sample preparation, buffer and mobile phase preparation, and sample dilution in LC-MS. Guarantees a contamination-free baseline, which is critical for sensitive detection [7]. |
| Leak-Tight Column Connectors | A common source of gas leaks in GC-MS; using high-quality, properly installed connectors is vital for maintaining system integrity, sensitivity, and accurate quantification [44]. |
| Certified Standard Solutions | Used for instrument calibration, method development, and quantification. Their purity and certification are fundamental for achieving accurate and legally defensible results [47]. |
| SPME Fibers & HPLC-Grade Solvents | Solid-phase microextraction (SPME) fibers are used for sample extraction and concentration (e.g., in HS-SPME-GC-MS). HPLC-grade solvents ensure clean baselines and consistent chromatographic performance [47] [45]. |
Quantitative Data on Techniques and Applications
The following table summarizes quantitative data and key application areas for prominent hybrid techniques, highlighting their specific strengths.
Table 2: Hybrid Technique Quantitative Data and Applications
| Technique | Key Performance Metric | Primary Application Areas |
|---|---|---|
| GC-MS [43] | High separation efficiency for volatile compounds. | Environmental monitoring, forensic analysis, aroma compound profiling [43] [47]. |
| LC-MS [43] | Superior for analyzing thermally unstable compounds. | Pharmaceutical development, proteomics, metabolomics, food safety [43] [46]. |
| Orbitrap MS [46] | Mass resolution >100,000 at m/z 35,000. | Detailed molecular characterization in proteomics and structural biology [46]. |
| GC-IR [43] | Effective identification of functional groups. | Petrochemical analysis (e.g., gasoline components), environmental contaminant identification [43]. |
| LC-NMR [43] | Provides detailed molecular structural information. | Drug discovery, natural product research, metabolomics [43]. |
| Spectroscopy Software Market [1] | USD 1.1 Billion (2024), CAGR of 9.1% (2025-2034). | Ubiquitous across all sectors using spectroscopic analysis, especially pharmaceuticals [1]. |
Experimental Protocols for Hybrid Analysis
Protocol: Food Quality Assessment via UHPLC-MS/MS and GC-MS
This protocol is adapted from food analysis research for profiling compounds like amino acids and aroma volatiles [47].
1. Sample Preparation:
- For UHPLC-MS/MS (Amino Acids): Homogenize the food sample (e.g., honey or beef). Perform a solid-liquid extraction using a suitable solvent like methanol or water. Centrifuge the mixture and filter the supernatant through a 0.2 µm membrane prior to analysis.
- For GC-MS (Volatile Aroma Compounds): Use Headspace Solid-Phase Microextraction (HS-SPME). Place the sample in a sealed vial and incubate at a controlled temperature. Expose a coated SPME fiber to the sample headspace to adsorb volatile compounds, then desorb the fiber directly in the GC injector.
2. Instrumental Analysis:
- UHPLC-MS/MS Conditions:
- Column: C18 reversed-phase column (e.g., 2.1 x 100 mm, 1.7 µm).
- Mobile Phase: (A) Water with 0.1% formic acid; (B) Acetonitrile with 0.1% formic acid.
- Gradient: Ramp from 5% B to 95% B over 10-15 minutes.
- Mass Spectrometer: Triple quadrupole MS operated in Multiple Reaction Monitoring (MRM) mode for high sensitivity and selective quantification [47].
- GC-MS Conditions:
- Column: Mid-polarity fused silica capillary column (e.g., 30 m x 0.25 mm ID, 0.25 µm film).
- Temperature Program: Ramp from 40°C (hold 2 min) to 250°C at 10°C/min.
- Mass Spectrometer: Electron ionization (EI) source at 70 eV; scan range m/z 40-450 [47].
3. Data Processing:
- Identify compounds by comparing mass spectra to reference libraries (NIST for GC-MS) and by matching retention times and fragmentation patterns with authentic standards.
- Use chemometric software tools (e.g., OPLS-DA) to build predictive models that correlate analyte profiles (e.g., amino acids) with qualities like geographic origin or aroma [47].
Protocol: Contaminant Screening in Food Matrices using HPLC-MS/MS
This method is designed for detecting trace-level contaminants like antimicrobials in complex food matrices [47].
1. Sample Extraction and Cleanup (QuEChERS):
- Homogenize the sample (e.g., lettuce).
- Extract with acetonitrile and partition salts (magnesium sulfate, sodium chloride).
- Perform a cleanup step using dispersive Solid-Phase Extraction (d-SPE) with sorbents like PSA and C18 to remove interfering matrix components.
2. Instrumental Analysis:
- HPLC-MS/MS Conditions (QTRAP MS):
- Column: C18 reversed-phase column.
- Mobile Phase: (A) Water and (B) Methanol, both with 5mM ammonium formate.
- Gradient: Optimize for the target analytes.
- Mass Spectrometer: Triple quadrupole-linear ion trap hybrid system (QTRAP). Use MRM for quantification and enhanced product ion (EPI) scans for confirmatory library matching.
- Validation: The method should be validated for parameters including specificity, linearity, recovery, precision, and limits of detection (LOD) and quantification (LOQ), which can be as low as 0.8 µg·kg⁻¹ for certain analytes [47].
Workflow Diagram for Systematic Troubleshooting
The following diagram outlines a logical, step-by-step workflow for diagnosing common issues in hybrid analytical systems, integrating checks for both chromatographic and detection components.
Systematic Troubleshooting Workflow
Advancements in Software and Data Analysis
The integration of advanced software is paramount for leveraging the full potential of hybrid techniques. Key trends include the incorporation of Artificial Intelligence (AI) and Machine Learning (ML) to enhance data processing speed, enable sophisticated pattern detection, and provide predictive analytics for spectral interpretation [1]. There is also a significant shift towards cloud-based and remotely accessible software solutions, which facilitate collaboration among geographically dispersed research teams and provide scalable computing resources for handling large spectral datasets [1].
The development of open-source software platforms, such as the Workbench for Imaging Spectroscopy Exploration and Research (WISER), addresses the need for flexible, modifiable analysis tools that can be customized for specific research requirements in imaging spectroscopy [37]. Furthermore, software is increasingly focusing on user accessibility, featuring intuitive dashboards, automated workflows, and customizable reporting to make powerful analytical tools available to a broader range of users, including non-specialists [1]. These advancements collectively make data analysis more efficient, collaborative, and accessible, directly supporting the complex data interpretation needs of researchers using hybrid MS, chromatography, and spectroscopy systems.
Technical Support Center: Troubleshooting Guides and FAQs
This section provides practical solutions for common issues encountered in FT-IR and QCL microscopy, directly supporting research in spectroscopy data analysis.
Frequently Asked Questions (FAQs)
Q: What is the core difference between FT-IR and QCL microscopy?
- A: FT-IR spectroscopy uses a broadband thermal source to collect a full infrared spectrum at once, offering a wide spectral range but requiring sensitive, often cooled detectors. QCL microscopy uses a tunable laser source that emits at specific wavelengths, providing a much higher spectral power density that enables faster imaging speeds and the use of room-temperature detectors, but over a more limited spectral range [48].
Q: Can FT-IR and QCL technologies be combined?
- A: Yes, modern instrumentation platforms like the HYPERION II now seamlessly integrate both FT-IR and QCL (Infrared Laser Imaging) in a single instrument. This allows researchers to leverage the full spectral range of FT-IR for initial analysis and then use the high speed of QCL for targeted chemical imaging [49].
Q: What are "coherence artefacts" in QCL imaging and how can they be mitigated?
- A: Coherence artefacts, such as fringes and speckles in IR images, are physical interference patterns caused by the highly coherent nature of laser light used in QCL systems. They can obscure chemical information. Bruker's ILIM technology addresses this with a patented hardware-based spatial coherence reduction method to collect artifact-free chemical images [49] [48].
Q: My FT-IR spectrum has strange, sharp negative peaks. What is the likely cause?
Q: Why does my spectrum of a plastic sample look different when I analyze the surface versus a freshly cut interior section?
- A: ATR is a surface-sensitive technique. Differences between surface and bulk spectra can be due to surface oxidation, migration of additives (like plasticizers) to or from the surface, or other effects from sample processing. Analyzing both surfaces provides valuable chemical insight into these heterogeneities [50].
Troubleshooting Common Experimental Issues
Problem 1: Noisy or Low-Intensity Spectra in FT-IR
- Potential Cause: Inadequate purging or contamination of the optical path.
- Solution: Ensure the instrument is properly purged with dry, CO₂-free air to minimize spectral contributions from atmospheric water vapor and CO₂. For instruments like the Bruker Vertex NEO, utilizing its vacuum optical path can effectively eliminate these interferences [7].
Problem 2: Distorted Peaks in Diffuse Reflection Measurements
- Potential Cause: Incorrect data processing.
- Solution: Spectra collected in diffuse reflection should be processed in Kubelka-Munk units, not absorbance. Converting to Kubelka-Munk provides a linear relationship between concentration and signal, correcting the distorted and saturated peaks that appear in the absorbance display [10] [50].
Problem 3: Unusual Spectral Features or "Ghost" Peaks
- Potential Cause: External instrument vibrations.
- Solution: FT-IR spectrometers are highly sensitive to physical disturbances. Ensure the instrument is placed on a stable, vibration-free bench, away from vacuum pumps, chillers, or other sources of vibration [10].
The 2025 Landscape of Novel Spectrometry Platforms
The global market for novel spectrometry platforms is experiencing robust growth, driven by demand from pharmaceutical, biotechnology, and applied industries.
Table 1: Global Novel Spectrometry Platforms Market Size and Growth [51] [52] [53]
| Metric | Value | Time Period / CAGR |
|---|---|---|
| 2024 Market Size | $4.39 - $14.52 billion | Base Year 2024 |
| 2025 Market Size | $4.76 - $15.81 billion | Forecasted |
| 2032/2034 Market Size | $6.47 - $26.20 billion | Forecasted |
| Compound Annual Growth Rate (CAGR) | 8.0% - 8.8% | 2024-2029/2032 |
Table 2: Novel Spectrometry Market Share by Spectrometer Type (2024) [52] [53]
| Spectrometer Type | Approximate Market Share |
|---|---|
| Atomic Absorption Spectrometer | 25.6% |
| Mass Spectrometer | Significant share (exact % varies by report) |
| Near Infrared Spectrometer | Significant share (exact % varies by report) |
| Nuclear Magnetic Resonance (NMR) Spectrometer | Significant share (exact % varies by report) |
| Raman Spectrometer | Significant share (exact % varies by report) |
| X-Ray Fluorescence Spectrometer | Significant share (exact % varies by report) |
Table 3: Regional Market Analysis (2024) [52]
| Region | Market Share | 2024 Market Size (USD Million) | Forecasted CAGR |
|---|---|---|---|
| North America | >40% | 5,807.28 | 7.0% |
| Europe | >30% | 4,355.46 | ~7.3% |
| Asia Pacific | ~23% | 3,339.19 | 10.8% |
| Latin America | >5% | 725.91 | 8.2% |
| Middle East & Africa | ~2% | 290.36 | 8.5% |
Key Market Trends and Drivers
The market's growth is propelled by several key factors:
- Pharmaceutical R&D: Increasing focus on drug discovery and personalized medicine demands advanced analytical tools for molecular characterization, impurity detection, and high-throughput screening [51] [53].
- Demand for High-Resolution Tools: Industries like food and beverage, environmental monitoring, and semiconductors require higher precision, sensitivity, and reproducibility for quality control and compliance [53].
- Technological Convergence: The integration of Artificial Intelligence (AI) and machine learning with spectroscopy software enhances data analysis, enabling faster processing, pattern recognition, and predictive analytics [1].
- Portability and Miniaturization: There is a significant trend towards developing portable and handheld spectrometers for on-site analysis in fields such as agriculture, forensics, and environmental monitoring [7] [53].
Experimental Protocols for Advanced Spectroscopic Analysis
Protocol: Combined FT-IR and QCL Microscopy for Heterogeneous Sample Analysis
This protocol utilizes the complementary strengths of FT-IR and QCL technologies for comprehensive sample characterization [49] [48].
1. Instrument Setup and Alignment
- Mount the sample on the microscope stage. For the HYPERION II platform, select an appropriate objective (e.g., 15x or 36x IR objective for transmission, or a 20x ATR objective for surface analysis).
- If using a liquid nitrogen-cooled MCT detector for FT-IR, ensure the detector is properly cooled. For QCL imaging, the room-temperature microbolometer array does not require cooling.
2. Initial FT-IR Survey Measurement
- Define a region of interest (ROI) on the sample using the visible camera.
- Acquire a high-quality FT-IR hyperspectral data cube (map or image) using a focal-plane array (FPA) detector. Parameters: typically 4 cm⁻¹ or 8 cm⁻¹ spectral resolution, 64-128 scans per spectrum to ensure a good signal-to-noise ratio.
- This step provides the full mid-IR spectrum (e.g., 4000 - 900 cm⁻¹) for every pixel in the ROI.
3. Data Analysis and QCL Wavelength Selection
- Process the FT-IR data cube using spectroscopy software (e.g., OPUS, WISER). Identify specific chemical components and their unique absorption bands (e.g., the carbonyl stretch at ~1720 cm⁻¹ for a specific polymer).
- Based on this analysis, select one or several characteristic wavenumbers for detailed, rapid imaging using the QCL source.
4. High-Speed QCL Imaging
- Switch the instrument to ILIM (Infrared Laser Imaging) mode.
- Configure the QCL to tune to the selected wavenumber(s). The patented coherence reduction hardware should be engaged to minimize artefacts.
- Acquire the chemical image. The QCL system can achieve imaging speeds up to 6.4 mm² per second at a single wavenumber, orders of magnitude faster than the FPA imaging in this targeted application.
5. Data Correlation and Validation
- Overlay the chemical images obtained from the QCL with the visual image and the FT-IR spectral data map.
- Use the full FT-IR spectrum as a reference to verify the chemical assignments made from the single-wavelength QCL image, ensuring robustness and reliability.
Protocol: ATR-FT-IR Analysis for Surface vs. Bulk Characterization
This protocol is designed to identify and analyze chemical differences between the surface and bulk of a material, such as a polymer film [50].
1. Sample Preparation
- Divide the sample into two portions.
- Leave the first portion "as received" for surface analysis.
- For the second portion, use a clean microtome blade or scalpel to make a fresh cut, exposing the bulk material.
2. Background Collection
- Clean the ATR crystal (diamond is common) with a suitable solvent (e.g., isopropanol) and wipe dry with a lint-free cloth.
- Collect a background spectrum with a clean, empty crystal. Parameters: 4 cm⁻¹ resolution, 16-32 scans.
3. Surface Spectrum Acquisition
- Place the "as received" sample on the ATR crystal and apply consistent pressure using the instrument's pressure clamp.
- Acquire the spectrum.
4. Bulk Spectrum Acquisition
- Remove the first sample and repeat the cleaning process for the ATR crystal.
- Place the freshly cut sample on the crystal, ensuring the new interior surface is in contact with the crystal.
- Acquire the spectrum using identical parameters.
5. Data Processing and Interpretation
- Apply an ATR correction algorithm to all spectra to account for the wavelength-dependent depth of penetration.
- Compare the surface and bulk spectra. Differences in peak ratios, the presence or absence of peaks (e.g., C=O stretch from oxidation, changes in plasticizer peaks), indicate chemical gradients within the material.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 4: Key Research Reagent Solutions for Spectroscopy
| Item / Reagent | Function / Application | Technical Notes |
|---|---|---|
| ATR Crystals (Diamond, ZnSe, Ge) | Enables surface-specific infrared analysis via attenuated total reflection. | Diamond is durable and chemically inert; ZnSe offers a good balance of performance and cost but is softer; Ge provides a shallow depth of penetration for strong absorbers [50]. |
| Ultrapure Water (e.g., from Milli-Q SQ2 systems) | Critical for sample preparation, dilution, and cleaning in sensitive analyses to prevent contamination. | Used in preparation of buffers, mobile phases, and for cleaning optics and accessories [7]. |
| Liquid Nitrogen (LN₂) | Cools Mercury Cadmium Telluride (MCT) detectors to reduce thermal noise for high-sensitivity FT-IR measurements. | Required for high-sensitivity FT-IR mapping and imaging with LN₂-cooled MCT detectors [49] [48]. |
| Solvents for Cleaning (e.g., Isopropanol, HPLC-grade Methanol) | Cleaning of ATR crystals, optical windows, and sample substrates to prevent spectral contamination. | Must be spectroscopic grade to avoid leaving residues. Essential for troubleshooting negative peaks in ATR spectra [10] [50]. |
| Microtome / Scalpel | Provides a fresh, clean cross-section of a sample to expose the bulk material for analysis. | Key for differentiating surface chemistry from bulk chemistry in materials science and failure analysis [50]. |
| Spectral Libraries & Software (e.g., WISER, OPUS) | Provides reference spectra for compound identification and tools for data processing, visualization, and analysis. | Open-source software like WISER supports analysis of imaging spectroscopy datasets, offering spatial/spectral subsetting, math toolkits, and plugin APIs [37]. |
Troubleshooting Guides
Incomplete Sample Digestion
Problem: Samples are not fully digested, leading to low analyte recovery and inaccurate results.
Solutions:
- Verify Method Parameters: Ensure temperature, pressure, and time are optimized for your sample matrix. Difficult samples may require Single Reaction Chamber (SRC) technology for more uniform digestion [54].
- Check Acid Compatibility: Confirm the acid mixture is appropriate for your sample type. Automating reagent dosing can improve consistency [54].
- Inspect Vessels: Look for signs of wear or damage in digestion vessels that could cause pressure loss [54].
Sample Contamination
Problem: Contaminants are introduced during preparation, skewing results.
Solutions:
- High-Purity Acids: Use sub-boiling distilled acids. Implementing in-house acid purification can reduce costs and contamination risk [54].
- Automated Cleaning: Use automated acid-steam cleaning for labware instead of manual washing to prevent cross-contamination [54].
- Clean Environment: Perform sample handling in controlled environments and use filter tips to minimize airborne contamination [55].
Low Laboratory Throughput
Problem: Sample preparation bottlenecks delay overall analysis.
Solutions:
- Workflow Analysis: Identify and optimize slowest steps, often reagent addition, vessel handling, or cleaning [54].
- Automate Dosing: Automated reagent addition increases speed and consistency [54].
- Parallel Processing: Use systems like simultaneous filtration (SFS-24) or SRC digestion to process multiple samples at once [54].
Inconsistent Results Between Replicates
Problem: High variability in results from identical samples.
Solutions:
- Standardize Techniques: Implement automated systems for reagent dosing and vessel handling to minimize operator variation [54].
- Monitor Sample Integrity: Track potential degradation by analyzing samples at each step using methods like Western Blot [55].
- Control Environment: Maintain consistent temperature (4°C during work, -20°C to -80°C for storage) and use protease inhibitors for unstable proteins [55].
Frequently Asked Questions (FAQs)
Workflow Strategy
Q: What is a 'total workflow' approach and why is it important? A: A 'total workflow' approach optimizes all steps in sample preparation—not just digestion—including acid purification, reagent dosing, vessel handling, and cleaning. This comprehensive view improves throughput, data quality, cost-effectiveness, and safety [54].
Q: How can I identify bottlenecks in my current workflow? A: Track time and resources for each step from sample receipt to analysis. Common bottlenecks include manual reagent addition, vessel handling, and cleaning. Addressing these through automation can yield significant improvements [54].
Technical Issues
Q: How can I prevent the loss of low-abundance proteins during preparation? A: Scale up your starting material, use fractionation to enrich low-abundance targets, and add protease inhibitors to buffers (ensure they are removed before trypsinization). Always monitor step-wise yield with controls [55].
Q: What are the signs of poor sample preparation in spectral analysis? A: In IR spectroscopy, broad peaks at 0% transmittance indicate overly thick samples. A sloping baseline suggests incomplete grinding for KBr pellets. Negative peaks often stem from a contaminated background scan [56].
Q: How do I choose between rotor-based and single reaction chamber (SRC) digestion? A: Rotor-based systems are "workhorses" but can be cumbersome. SRC provides higher throughput, faster digestion of difficult samples, and reduced labor through parallel processing of different sample types under uniform conditions [54].
Workflow Optimization Data
Spectroscopy Software Market Trends
Table: Key Market Trends and Drivers in Spectroscopy Software
| Trend Category | Specific Trend | Impact on Elemental Analysis |
|---|---|---|
| Technological | Integration of AI and ML | Enhances data processing, pattern recognition, and predictive analytics [1] |
| Deployment | Growth of cloud-based platforms | Facilitates remote collaboration and data sharing among teams [1] |
| Usability | Development of intuitive dashboards | Makes software accessible to non-specialists, speeding up adoption [1] |
| Application | Increased use in pharmaceuticals | Drives demand for software in drug discovery and quality control [1] |
| Regulatory | Compliance with food/environmental safety | Ensures software meets quality and regulatory standards [1] |
Table: Global Spectroscopy Software Market Metrics (2024-2034)
| Metric | 2024 Value | Projected 2034 Value | CAGR (2025-2034) |
|---|---|---|---|
| Market Size | USD 1.1 Billion [1] | USD 2.5 Billion [1] | 9.1% [1] |
| Pharmaceutical Segment Share | 28.9% [1] | - | - |
| On-Premises Deployment Segment | USD 549.5 Million [1] | - | - |
Experimental Protocols
'Total Workflow' Optimization Protocol
Objective: Implement and validate a complete sample preparation workflow for elemental analysis that maximizes throughput, data quality, and safety.
Materials:
- Samples for analysis
- Milestone ultraWAVE SRC microwave digestion system (or equivalent) [54]
- Automated reagent dosing system (e.g., easyFILL) [54]
- In-house acid purification system (sub-boiling distillation) [54]
- Automated labware cleaner (acid-steam system) [54]
- Simultaneous Filtration System (SFS-24) [54]
- High-purity acids and reagents
Procedure:
- Acid Purification: Purify acids in-house using sub-boiling distillation to ensure quality and reduce costs [54].
- Automated Reagent Addition: Use an automated dosing system to add consistent acid volumes to digestion vessels, improving reproducibility and safety [54].
- Microwave Digestion: Digest samples using Single Reaction Chamber (SRC) technology. The ultraWAVE 3 allows simultaneous processing of different sample types under uniform conditions (increased temperature and pressure) for complete digestion [54].
- Sample Filtration: Following digestion, filter samples using the SFS-24 simultaneous system to remove particulates and prepare clear solutions for analysis [54].
- Automated Cleaning: Clean all digestion vessels and labware using an automated acid-steam system to prevent cross-contamination and ensure readiness for the next run [54].
Validation:
- Compare digestion efficiency of the optimized workflow against traditional methods by measuring analyte recovery for certified reference materials.
- Monitor sample throughput (samples per day) and operational costs before and after implementation.
- Assess reproducibility by calculating the relative standard deviation (RSD) of replicate measurements.
Workflow Visualization
Diagram Title: Optimized Total Workflow for Elemental Analysis
The Scientist's Toolkit: Key Research Reagent Solutions
Table: Essential Materials and Equipment for an Optimized Workflow
| Item | Function | Application Notes |
|---|---|---|
| In-house Acid Purification System | Produces high-purity acids via sub-boiling distillation, reducing cost and contamination risk [54]. | Critical for trace element analysis. Ensures supply chain resilience. |
| Automated Reagent Dosing System (e.g., easyFILL) | Precisely adds concentrated acids, improving consistency and enhancing operator safety [54]. | Eliminates manual handling errors and exposure to fumes. |
| Single Reaction Chamber (SRC) Digester | Digests multiple samples simultaneously under uniform high temperature/pressure, even for difficult matrices [54]. | Increases throughput and eliminates incomplete digestion issues. |
| Simultaneous Filtration System (SFS-24) | Filters multiple digested samples in parallel under vacuum, saving time and fume hood space [54]. | Uses inexpensive, solvent-compatible funnels. |
| Automated Acid-Steam Cleaner | Cleans digestion vessels and labware automatically, preventing cross-contamination and saving labor [54]. | More efficient and consistent than manual cleaning. |
| Protease Inhibitor Cocktails (EDTA-free) | Prevents protein degradation during sample preparation steps, preserving analyte integrity [55]. | Must be removed before trypsinization steps. |
| Filter Tips & HPLC-Grade Water | Prevents introduction of contaminants like keratin or polymers that interfere with sensitive detection [55]. | Essential for low-abundance analyte analysis. |
Troubleshooting Guides and FAQs
A-TEEM Spectroscopy
Frequently Asked Questions
Q1: Our A-TEEM data shows non-linear fluorescence response at higher concentrations, skewing quantitative models. What is the cause and solution?
A: This is a classic symptom of the Inner Filter Effect (IFE), a common issue in fluorescence spectroscopy where the sample absorbs both the excitation light and the emitted fluorescence, leading to reduced and distorted signals [57] [58]. The A-TEEM technology is specifically designed to correct for this.
- Root Cause: In samples with high absorbance, the excitation light intensity is not constant throughout the sample pathlength. The fluorescence emitted from deeper within the sample can also be re-absorbed before it is detected [58].
- Solution: The key advantage of A-TEEM is the simultaneous measurement of absorbance and fluorescence [57]. Use the instrument's software to apply an internal IFE correction. This uses the simultaneously acquired absorbance spectrum to mathematically correct the fluorescence EEM, resulting in a concentration-independent molecular fingerprint suitable for robust quantitative analysis [57] [58].
Q2: We need to deploy an A-TEEM method for GMP batch release testing. What software and documentation are required for regulatory compliance?
A: Transitioning to a GMP environment requires specific software features and validation documentation.
- Required Solution: Implement the A-TEEM Compliance Package [59]. This package is designed to meet the stringent requirements of GMP laboratories.
- Key Components:
- IQ/OQ Documentation: Installation and Operational Qualification documentation to verify and validate that the instrument is installed correctly and operates according to specifications [59].
- 21 CFR Part 11 Compliant Software: Software that ensures electronic records and signatures are trustworthy, reliable, and equivalent to paper records [58]. This includes features like audit trails and user access controls.
- Guided Workflow Software: Use multivariate analysis software like A-TEEM Direktor, which provides a guided workflow to standardize the analysis process and minimize user-induced variability [59].
- PAT Software Integration: The EzPAT OPC UA Server enables Process Analytical Technology (PAT) software to control A-TEEM acquisitions, facilitating integration into automated manufacturing processes [59].
Q3: How can we rapidly distinguish between highly similar molecules, such as isomers, using A-TEEM?
A: A-TEEM excels at this by generating highly specific molecular fingerprints.
- Methodology: The technique combines absorbance data with fluorescence EEMs, capturing subtle variations in the molecular structure and micro-environment that other techniques might miss [57].
- Example: Cresol isomers (ortho-, meta-, para-cresol), which have nearly identical structures, can be rapidly resolved. Their unique, concentration-independent A-TEEM fingerprints, when coupled with multivariate analysis, allow for clear identification and quantification. Data acquisition for this is rapid, taking only seconds [57].
Troubleshooting Guide: Poor Model Performance in A-TEEM Multivariate Analysis
| Symptom | Potential Cause | Recommended Action |
|---|---|---|
| Low classification/quantification accuracy in model [57] | Insufficient selectivity in the fingerprint data. | Utilize the multi-block organization of both absorbance and fluorescence data. The combination significantly enhances the statistical significance of models by including data from both fluorescing and weakly- or non-fluorescing compounds [57] [58]. |
| Model fails to generalize to new samples [57] | Inner Filter Effects not corrected, making fingerprints concentration-dependent. | Ensure the IFE correction is applied during data pre-processing to generate true concentration-independent molecular fingerprints [57]. |
| Inability to track process in real-time [57] | Measurement speed is too slow. | Leverage the CCD detector of instruments like the Aqualog for rapid data acquisition, enabling measurements in seconds for real-time Process Analysis and Control [57]. |
Raman Spectroscopy
Frequently Asked Questions
Q1: Our Raman classification model, trained last month, now performs poorly on the same instrument. What could be causing this drift?
A: This is a recognized challenge related to the long-term instability of Raman setups. Device components can drift over time, leading to subtle changes in spectral intensity and wavenumber position [60].
- Root Cause: Instrumental variations over time are often random and can be caused by factors like laser power fluctuation, alignment changes, or environmental conditions [60].
- Solution:
- Implement a Weekly QC Protocol: Regularly measure stable standard references (e.g., Paracetamol EP, Polystyrene, Silicon). This monitors instrumental performance [60].
- Computational Correction: Employ advanced data processing methods. A study demonstrated that variations can be estimated using a Variational Autoencoder (VAE) and suppressed using the Extended Multiplicative Scattering Correction (EMSC) method, which improved prediction accuracy across different measurement days [60].
- Regular Wavenumber Calibration: Use standard materials like cyclohexane or paracetamol to perform consistent wavenumber calibration, ensuring peak positions remain stable [60].
Q2: When should we choose Raman spectroscopy over A-TEEM for biopharmaceutical analysis?
A: The choice depends on the analytical goal, as these techniques are often complementary [61].
The table below compares their core strengths to guide your selection:
| Feature | A-TEEM Spectroscopy | Raman Spectroscopy |
|---|---|---|
| Primary Strength | High-sensitivity fingerprinting of fluorescent/colored compounds [57] | Molecular structure and polymorph identification [61] |
| Key Application | Quantifying AAV empty/full capsids, monitoring cell media quality, vaccine fingerprinting [62] [57] [58] | Protein secondary structure analysis, contaminant identification, polymorph screening [62] [61] |
| Sensitivity | Excellent (ppb range for certain analytes) [57] | Less sensitive than A-TEEM; generally requires higher concentrations [57] |
| Water Interference | Insensitive to water, ideal for aqueous solutions [57] | Susceptible to interference from water [61] |
| Speed | Very fast (seconds per measurement) [57] | Fast (seconds to minutes) [61] |
Troubleshooting Guide: Addressing Common Raman Instrumental Issues
| Symptom | Potential Cause | Recommended Action |
|---|---|---|
| Weak or noisy signal [61] | Inherently weak Raman scattering signal; suboptimal instrument settings. | Increase integration time (e.g., 1 second used in stability study [60]), ensure laser is properly focused and aligned, and verify laser power output. |
| Spectral congestion/overlapping peaks [61] | Complex sample matrix with multiple components. | Apply multivariate curve resolution (MCR) or principal component analysis (PCA) to deconvolute overlapping signals [60]. |
| Poor model transfer between instruments [60] | Substantial device-to-device variation. | Perform rigorous calibration with standards, and apply warping or EMSC algorithms to align spectral features from different sources [60]. |
The Scientist's Toolkit: Essential Research Reagent Solutions
The following reagents are critical for system qualification, performance monitoring, and developing robust analytical methods.
Raman Spectroscopy QC Reagents
| Reagent/Item | Function | Key Note |
|---|---|---|
| Silicon Wafer | Intensity Calibration | Used to calibrate and monitor the intensity stability of the system using its sharp band at 520 cm⁻¹ [60]. |
| Cyclohexane | Wavenumber Calibration | A standard reference material with well-defined peaks for accurate wavenumber calibration [60]. |
| Paracetamol (EP) | Stability Benchmarking | A stable solid standard used to monitor the long-term reproducibility and focus stability of the system [60]. |
| Polystyrene | System Suitability Check | Provides a characteristic Raman spectrum to verify overall system performance and resolution [60]. |
A-TEEM Spectroscopy Application Standards
| Reagent/Item | Function | Key Note |
|---|---|---|
| Cresol Isomers | Method Selectivity Validation | Used to demonstrate the technique's capability to resolve highly similar molecular structures [57]. |
| Tryptophan/Tyrosine | Biomolecule Characterization | Used for characterizing proteins and their conformational changes, as these amino acids are highly fluorescent and environmentally sensitive [57]. |
| NAD(P)H | Bioprocess Monitoring | A key metabolic coenzyme; its A-TEEM fingerprint can be used to monitor cell viability and metabolic state in bioreactors [57]. |
Experimental Protocols & Workflows
Detailed Methodology: A-TEEM for Vaccine Fingerprinting and Rapid Identity Testing
This protocol is adapted from applications where A-TEEM replaces a 30-day potency test by providing a unique spectral fingerprint for final product verification [58].
Objective: To rapidly confirm the identity of a vaccine product prior to batch release.
Sample Preparation:
- Obtain a small aliquot of the final vaccine product.
- Gently solubilize or dilute the vaccine according to a standardized protocol to achieve an absorbance within the instrument's linear range (typically below ~2 AU) [57].
- Transfer the prepared sample to a suitable quartz cuvette.
Instrumentation and Software:
- Instrument: HORIBA Aqualog or similar A-TEEM spectrometer.
- Software: Instrument control software with IFE correction capabilities. Multivariate analysis software (e.g., A-TEEM Direktor, Solo) for model building and prediction [59] [57].
- Accessory (Optional): FAST-01 Autosampler for automated, temperature-controlled batch analysis [58].
Procedure:
- Data Acquisition: Load the sample and acquire the A-TEEM data. The simultaneous absorbance and fluorescence EEM data are collected in seconds [57].
- Data Pre-processing: Apply the internal IFE correction algorithm to generate a concentration-independent molecular fingerprint [57].
- Model Application: Input the corrected fingerprint into a pre-validated multivariate classification model (e.g., PCA-LDA, or a multi-block model). This model is built using a library of fingerprints from all approved vaccine products.
- Result Interpretation: The model will assign the unknown sample to a known vaccine identity with a defined confidence score. A match confirms product identity.
Detailed Methodology: Monitoring Long-Term Raman Instrument Stability
This protocol is based on a comprehensive study that systematically assessed a Raman device's performance over 10 months [60].
Objective: To systematically track and quantify the long-term performance drift of a Raman spectrometer.
QC Reference Materials:
- Select stable substances covering a range of molecular features. The study used [60]:
- Solvents: Ethanol, Isopropanol, DMSO, Benzonitrile.
- Carbohydrates: Fructose, Glucose, Sucrose.
- Lipids: Squalene.
- Standards: Paracetamol, Polystyrene.
Instrumentation and Data Analysis:
- Instrument: A Raman spectrometer (e.g., HTS-RS system with a 785 nm laser) [60].
- Software: Data analysis pipeline capable of PCA, k-means clustering, classification, and advanced algorithms like VAE and EMSC [60].
Procedure:
- Weekly Measurement: On a scheduled day each week, acquire approximately 50 spectra from each of the QC reference materials [60].
- Routine Pre-processing: Process all spectra using a standard pipeline: despiking, wavenumber calibration, baseline correction, and vector normalization [60].
- Stability Benchmarking:
- Correlation Analysis: Calculate the Pearson's Correlation Coefficient (PCC) between the mean spectra of different measurement days for each substance. A decrease in PCC indicates spectral drift [60].
- Clustering Analysis: Perform k-means clustering on data from all days. Ideal stability is indicated when spectra cluster by substance rather than by measurement day [60].
- Classification Accuracy: Train a classifier (e.g., on Day 1 data) and test its performance on data from subsequent days. A drop in accuracy indicates instrumental drift affecting predictive models [60].
- Corrective Action: If significant drift is detected, apply computational corrections (e.g., EMSC) to the data or schedule instrumental maintenance and re-calibration [60].
Troubleshooting and Optimization: Ensuring Data Integrity and Instrument Performance
Troubleshooting Guides
Guide 1: Vacuum System Failures
Problem: The spectrometer's vacuum pump cannot achieve or maintain the required vacuum level, leading to poor performance, especially for elements like carbon, phosphorus, and sulfur that require analysis in the short ultraviolet spectral region [63].
Diagnosis and Solutions: Perform the following checks to diagnose the issue systematically.
Step 1: Check for System Leaks Close the vacuum valve. If the vacuum level is maintained well, the issue likely lies with the vacuum probe or gauge. If the vacuum level rapidly decreases, there is a leak in the vacuum chamber [63].
- Solution for Leaks: Open the chamber and reseal all components. Key areas to check include [63]:
- The grating cover 'O'-ring.
- The 'O'-ring and quartz glass of the incident window (ensure correct orientation).
- The screw connection and sealing ring for the incident slit.
- The socket holder for the photomultiplier tube's high-voltage supply cable. After resealing, close the chamber and pump for 30-40 minutes. The vacuum should approach an acceptable level (e.g., near 100 Torr for some systems). If not, resealing is required [63].
- Solution for Leaks: Open the chamber and reseal all components. Key areas to check include [63]:
Step 2: Verify Vacuum Probe and Gauge If the chamber is sealed but the gauge shows a poor vacuum (e.g., 20-30 Torr), the vacuum probe's thermistor may have failed [63].
- Solution: Replace the vacuum probe. Alternatively, for some systems (like the DV-4), fine-tuning the resistor on the vacuum gauge to rebalance the bridge circuit can restore operation [63].
Step 3: Inspect the Vacuum Pump and Fluid Continuous pumping for many hours without results can indicate a saturated pump fluid [63].
- Solution: Check the vacuum pump oil. If the oil surface shows water vapor, the collector's molecular sieve may be wet and require heating to dry it out [63]. For liquid ring vacuum pumps, also ensure the water supply is within the normal specified range, as an inadequate supply can cause poor vacuum performance [64].
Step 4: Examine Pump Mechanics For mechanical pumps, internal wear can cause a loss of vacuum.
- Solution: Reduce the clearance between the impeller and the distribution board to between 0.15-0.20mm. Inspect the mechanical seal for damage and replace it if necessary, as damage can cause air leakage [64].
Preventive Maintenance:
- Routine Inspection: Conduct regular inspections of the pump's operation and lubrication [64].
- Seal and Bearing Care: Regularly inspect mechanical seals for wear and monitor bearing clearance [64].
- Optimize Parameters: Maintain the vacuum level, suction pressure, and discharge pressure within the manufacturer's recommended ranges [64].
Guide 2: Optical Component Contamination
Problem: Reduced sensitivity, inaccurate results, or poor ionization efficiency due to contamination of optical components or the ionization source [65].
Diagnosis and Solutions:
Area 1: Ionization Source Contamination Contamination from sample residue, solvent deposits, or atmospheric gases can coat the ionization source [65].
- Symptoms: Poor ionization efficiency, reduced sensitivity, signal instability [65].
- Solution:
- Regular Inspection: Visually inspect the ionization source periodically for buildup [65].
- Cleaning: Follow the manufacturer's instructions for cleaning. Use recommended cleaning solutions and procedures to remove contaminants without damaging components [65].
- Alignment: Verify the source alignment after cleaning, as misalignment can also cause reduced sensitivity [65].
Area 2: Nebulizer and Sample Introduction System Blockage The sample introduction system is critical and prone to blockage or wear, especially with complex matrices [66].
- Symptoms: Erratic spray pattern, reduced signal intensity, poor precision [66].
- Solution:
- Visual Inspection: Aspirate water to check for an erratic aerosol pattern [66].
- Clearing Blockages: Remove blockages by applying backpressure with an argon line or by immersing the nebulizer in an appropriate acid or solvent. An ultrasonic bath can aid dissolution, but check with the manufacturer first. Never use wires to clear the tip, as this can cause permanent damage [66].
- Use Specialized Tools: Consider using a digital thermoelectric flow meter to monitor sample uptake or a commercial nebulizer-cleaning device that safely dislodges particles with pressurized cleanser [66].
Area 3: Optical Fiber and Probe Degradation Fiber optic cables and probes used for light transmission are susceptible to damage and contamination [67].
- Symptoms: Significant light loss, broken fibers, scratches affecting transmission [67].
- Solution and Care:
- Handle with Care: Always use one hand to hold the fiber connector and the other to remove end caps to avoid strain [67].
- Mind the Bend Radius: Avoid sharp bends. The table below provides guidelines for different fiber types [67].
- Avoid Excessive Heat: Keep fibers away from high-temperature sources. Standard fiber jacketing can melt if exposed to excessive heat [67].
- Clean and Protect: When not in use, replace end caps. Clean fiber ends periodically with lens paper and a solvent like distilled water, alcohol, or acetone [67].
Optical Fiber Bend Radius Guidelines [67]
| Fiber Core Size | Fiber Types | Long-Term Bend Radius (Storage) | Short-Term Bend Radius (In Use) |
|---|---|---|---|
| 50 ± 5 μm | VIS-NIR, UV-VIS | 4 cm | 2 cm |
| 100 ± 3 μm | VIS-NIR, UV-VIS | 4 cm | 2 cm |
| 200 ± 4 μm | VIS-NIR, UV-VIS, SR | 8 cm | 4 cm |
| 400 ± 8 μm | VIS-NIR, UV-VIS, SR | 16 cm | 8 cm |
| 600 ± 10 μm | VIS-NIR, UV-VIS, SR | 24 cm | 12 cm |
| 1000 ± 3 µm | VIS-NIR | 30 cm | 15 cm |
Frequently Asked Questions (FAQs)
Q1: What are the most common signs of a failing vacuum pump in a spectrometer? The most common signs are the inability to achieve or maintain the required vacuum level, excessive noise or vibration, and overheating. This failure directly impacts data quality, particularly for elements like carbon, phosphorus, and sulfur analyzed in the short UV range [63] [64].
Q2: How often should I clean the ionization source and nebulizer? The frequency depends on your sample workload and the types of samples analyzed. Nebulizers should be inspected every 1–2 weeks [66]. For the ionization source, refer to the manufacturer's instructions, but regular inspection is key. Laboratories with high sample throughput or those running corrosive samples will require more frequent cleaning [65] [66].
Q3: Can damaged optical fibers be repaired, or do they need to be replaced? Optical fibers with broken cores or severely damaged connectors typically need to be replaced. Damage from excessive bending or kinking causes permanent light loss. Proper handling and adherence to bend radius guidelines are crucial for prevention [67].
Q4: How does spectrometer software aid in troubleshooting these issues? Modern spectroscopy software is increasingly incorporating AI and machine learning to improve data analysis, pattern detection, and predictive analytics [1]. Furthermore, software can provide real-time monitoring of instrument parameters, such as vacuum levels and signal stability, alerting users to performance deviations that may indicate developing hardware issues. Platforms like the open-source WISER software also provide modular toolkits for in-depth data interrogation, which can help identify anomalies linked to specific hardware problems [37].
Experimental Protocols for System Verification
Protocol 1: Vacuum Integrity Test
This protocol provides a step-by-step methodology to verify the integrity of the spectrometer's vacuum system, a critical pre-experiment check.
1. Objective: To determine if the spectrometer's vacuum chamber and pumps are functioning correctly and holding a seal.
2. Materials:
- Spectrometer with vacuum system
- Leak detector (compatible with the instrument)
- Manufacturer-approved vacuum grease
3. Methodology:
- Initial Pump Down: Initiate the vacuum pump and record the time taken to reach the operational vacuum level specified in the manufacturer's manual.
- Isolation Test: Once the operational vacuum is reached, close the valve isolating the vacuum chamber from the pump. Monitor the vacuum gauge for a set period (e.g., 30 minutes).
- Leak Detection: If a significant pressure rise is observed during the isolation test, use a leak detector to systematically check all potential leak points. These include [65] [63]:
- Connections and fittings between the pump and chamber.
- Seals and gaskets, especially around the grating cover, incident window, and access ports.
- The vacuum chamber housing itself.
- Inspection and Re-sealing: If a leak is identified at a seal, carefully disassemble the connection, clean the old grease, and reapply a thin layer of manufacturer-approved vacuum grease before resealing [63].
4. Data Analysis: A stable vacuum reading during the isolation test indicates good system integrity. A rising pressure indicates a leak that must be located and sealed before proceeding with analyses.
Protocol 2: Optical Path Performance Verification
This protocol verifies the performance and cleanliness of the optical path, including the source and light transmission components.
1. Objective: To confirm that the ionization source and optical components are clean, aligned, and functioning optimally.
2. Materials:
- Spectrometer system
- Certified standard reference material
- Lens paper and HPLC-grade methanol
- Digital thermoelectric flow meter (optional)
3. Methodology:
- Baseline Signal Measurement: Introduce a pure solvent or blank matrix into the sample introduction system and acquire a spectral baseline. Note the signal intensity and noise level.
- Standard Analysis: Analyze a certified standard reference material with a known concentration. Record the signal intensity, stability, and calculated concentration.
- Nebulizer Inspection: Aspirate pure water and visually inspect the aerosol pattern from the nebulizer for consistency and the absence of large droplets [66].
- Flow Rate Check (Optional): Use a digital thermoelectric flow meter inline to verify the actual sample uptake rate matches the set parameter [66].
- Source and Fiber Inspection: If accessible and following safe shutdown procedures, visually inspect the ionization source for contamination [65]. For fiber-optic systems, inspect the connector ends for scratches or dirt and clean with lens paper and methanol if needed [67].
4. Data Analysis: Compare the signal intensity and stability of the standard against historical data from the same instrument. A significant drop in sensitivity or an increase in noise suggests contamination (e.g., at the ionization source) or a blockage (e.g., in the nebulizer). An erratic aerosol pattern confirms a nebulizer issue.
Diagnostic Workflows and System Relationships
Vacuum Pump Failure Diagnosis
Optical Contamination Diagnosis
The Scientist's Toolkit: Research Reagent Solutions
The following table details key consumables and materials essential for the maintenance and troubleshooting of spectrometers, as cited in the experimental protocols above.
| Item | Function in Spectrometer Maintenance |
|---|---|
| Leak Detector | Used to identify and locate minute leaks in the vacuum system, which are critical for resolving pump-down failures [65]. |
| Vacuum Grease | A high-purity, manufacturer-approved grease used to create airtight seals on 'O'-rings and flanges in the vacuum chamber [63]. |
| Digital Thermoelectric Flow Meter | A diagnostic tool placed inline to verify the actual sample uptake rate to the nebulizer, helping to identify blocked nebulizers or worn peristaltic pump tubing [66]. |
| Nebulizer-Cleaning Device | A specialized device that safely delivers a pressurized cleanser through the nebulizer capillary to dislodge particle build-up without causing damage [66]. |
| Lens Paper & HPLC-Grade Solvents | Used to gently clean the ends of optical fiber connectors and other sensitive optical surfaces without scratching or leaving residues [67]. |
| Certified Standard Reference Material | A sample with a known, certified composition used to verify the sensitivity, accuracy, and overall performance of the spectrometer after maintenance [66]. |
Frequently Asked Questions (FAQs)
Q1: My spectroscopic results are inconsistent between different instruments or runs. What could be the cause? Inconsistent results often stem from unwanted technical variation rather than true biological or chemical changes. Common sources include:
- Instrument Batch Effects: Differences between spectrometers can introduce systematic errors in measured biomarker concentrations [68].
- Drift Over Time: Analyte concentrations can shift due to instrument performance changes over time within the same spectrometer [68].
- Sample Handling Artefacts: The position of a sample on a 96-well plate (row or column) can affect readings due to intra-plate variation. The time between sample preparation and measurement can also lead to degradation and ongoing metabolism in the sample [68].
- Incorrect Data Pre-processing: Applying pre-processing techniques (like smoothing or scatter correction) without a clear justification, or applying them in the wrong order, can distort data and introduce errors [69].
Q2: What is the difference between data integrity and data quality? While related, these are distinct concepts crucial for reliable data:
- Data Integrity focuses on whether data are complete, consistent, accurate, trustworthy, and reliable throughout their life cycle. It ensures the data has not been altered or falsified and that the analytical process is traceable [70].
- Data Quality focuses on whether the data is fit for its intended purpose. This includes its precision, accuracy, format, and the supporting information, ensuring it is suitable for making scientific decisions [70]. Data integrity is a foundational requirement for data quality.
Q3: How can I improve the accuracy of my metabolite identification? Relying on a single analytical technique often leads to misidentified or unidentified metabolites [71]. To improve accuracy:
- Combine Complementary Techniques: Using both NMR and Mass Spectrometry (MS) is highly effective. NMR provides superior quantitative capabilities and structural information, while MS offers higher sensitivity and resolution. Together, they provide confirmatory evidence for metabolite identification [71].
- Utilize Multivariate Data Analysis (MVDA): MVDA is a powerful tool for building robust calibration models that correlate spectral data with reference analyte data, improving prediction accuracy [72].
Q4: My model's predictions are unreliable. How can I make them more robust? Unreliable models are often built on poor-quality data or use suboptimal pre-processing.
- Use Design of Experiments (DOE): Instead of relying on random process variations, a DOE approach allows you to proactively create a large design space with widespread process trajectories. This generates statistically relevant data for building more robust and reliable calibration models [72].
- Systematize Pre-processing: Use a systematic approach, like an experimental design, to test different combinations and orders of pre-processing steps (baseline correction, scatter correction, smoothing, scaling) to find the optimal strategy for your specific dataset [69].
Troubleshooting Guides
Problem 1: Inconsistent or Noisy Spectral Data
| Potential Cause | Recommended Action | Preventive Measure |
|---|---|---|
| Sub-optimal Signal-to-Noise | Evaluate if you can increase sample amount or the number of averaged scans [69]. | Prior to analysis, define the required resolution and signal-to-noise ratio based on the narrowest peak and the dynamic change of your system [69]. |
| Inappropriate Data Smoothing | Avoid applying smoothing filters (e.g., Savitzky-Golay) by default. Use them only when noise reduction is necessary and justify the parameters [69]. | Understand that smoothing can distort data. Use a systematic DOE approach to determine if smoothing improves your model's predictive error [69]. |
| Stray Light | Test for stray light, particularly at the ends of the instrument's spectral range, as it can cause significant photometric errors [73]. | Regularly maintain and calibrate the instrument using recommended standards [73]. |
Problem 2: Inaccurate Quantification or Concentration Prediction
| Potential Cause | Recommended Action | Preventive Measure |
|---|---|---|
| Technical Variation (Batch, Drift, Plate Effects) | Perform a rigorous quality control pipeline to remove unwanted variation. This involves regressing out effects of sample degradation time, plate row/column position, and drift over time [68]. | Implement standard operating procedures (SOPs) for sample handling, plating, and instrument calibration. Randomize sample placement where possible. |
| Poor Calibration Model | Use Analyte Spiking. Spiking with known concentrations of analytes breaks correlations between analytes and extends the concentration range, preventing cross-sensitivity and creating a more robust model [72]. | Build models using a Design of Experiments (DOE) approach to ensure your data covers a wide and realistic range of process variations [72]. |
| Incorrect Wavelength Calibration | Check the wavelength accuracy of your instrument using emission lines (e.g., Deuterium) or absorption bands from certified reference materials (e.g., Holmium oxide solution) [73]. | Follow a regular instrument qualification and calibration schedule as per the manufacturer's and regulatory guidelines [74]. |
Problem 3: Peaks are Misidentified or Overlooked
| Potential Cause | Recommended Action | Preventive Measure |
|---|---|---|
| Limited Analytical Platform | Confirm tentative identifications from one technique (e.g., MS) with a complementary technique (e.g., NMR). The combined evidence greatly increases confidence [71]. | Design studies to incorporate multiple analytical platforms from the start for more comprehensive metabolome coverage [71]. |
| Incorrect Baseline or Scatter Effects | Apply appropriate pre-processing techniques like Multiplicative Scatter Correction (MSC) or derivative filters to correct for baseline offsets and light scattering effects [69]. | Systematically test different pre-processing methods and their parameters to find the best combination for your specific data and analytical goal [69]. |
Experimental Protocols for Key Procedures
Protocol 1: A Rigorous QC Pipeline for Removing Technical Variation from Large-Scale NMR Data
This protocol, derived from the UK Biobank study, provides a method to systematically remove unwanted technical noise from large biomarker datasets [68].
- Log Transformation: Transform the original biomarker concentrations to a log scale. Apply a small offset to biomarkers with zero concentrations.
- Regress Out Sample Degradation Time: Perform a robust linear regression of the log-transformed data on the log of the time between sample preparation and measurement. Use the residuals for the next step.
- Adjust for Intra-Plate Variation: Sequentially regress the residuals from the previous step on plate row (categorical: A-H) and then on plate column (categorical: 1-12).
- Remove Inter-Plate Drift: Bin plates into groups by measurement date within each spectrometer. Regress the current residuals on this "date bin" factor as a categorical variable.
- Rescale to Absolute Concentrations: Transform the final residuals back to absolute concentrations by rescaling their distribution to match the original data's distribution.
- Remove Outlier Plates: Systematically identify and remove entire outlier plates that show strong, non-biological deviations, as these cannot be adjusted for.
The following workflow diagram illustrates this multi-step process:
Protocol 2: A Systematic Workflow for Spectroscopic Data Pre-processing
This protocol outlines a decision-making process for applying data pre-processing, helping to avoid the "black magic" of standard, unjustified workflows [69].
- Define Data Quality Issue: Identify the specific artefact in your raw data (e.g., baseline offset, scatter, noise).
- Formulate Pre-processing Experiments: Create a experimental design table (full factorial) that includes relevant techniques (Baseline, Scatter, Smoothing, Scaling) at different levels (e.g., "Yes"/"No").
- Execute and Evaluate: Run the pre-processing combinations from your design on a representative dataset.
- Select Optimal Strategy: Use a response variable like the Root-Mean-Square Error of Prediction (RMSEP) to identify the pre-processing strategy (and its order) that gives the best model performance.
- Implement and Document: Apply the optimal pre-processing workflow to the full dataset and thoroughly document all steps and parameters for reproducibility.
The logical flow of this protocol is shown below:
The Scientist's Toolkit: Essential Research Reagents & Materials
The following table details key materials and software tools essential for ensuring data quality in spectroscopic analysis.
| Item Name | Function / Purpose |
|---|---|
| Holmium Oxide (HoO) Solution | A certified reference material used to verify the wavelength accuracy of spectrophotometers across UV-Vis regions by checking its characteristic absorption bands [73]. |
| Deuterium Lamp | An emission line source used for high-precision wavelength calibration of spectrophotometers, especially in the UV region [73]. |
| Neutral Density Filters / Stray Light Filters | Solid or liquid filters used to test the photometric linearity and measure the level of stray light in a spectrophotometer, which is a critical parameter for accuracy [73]. |
| Multivariate Data Analysis (MVDA) Software | Software used to analyze complex, multivariate spectral data. It is essential for building robust calibration models that correlate spectral signals to reference analyte concentrations [72]. |
| Design of Experiments (DOE) Software | Software used to plan efficient and statistically sound experiments. It helps generate optimal datasets for building predictive models by systematically varying process parameters [72]. |
| Process Analytical Technology (PAT) Software | Integrated software that automates data collection from inline spectrometers (e.g., Raman) and enables real-time process monitoring and control in biomanufacturing [72]. |
Troubleshooting Guides
Problem: High Background Noise in XPS Analysis
Question: Why is my XPS spectrum showing unusually high carbon and oxygen backgrounds despite using high-purity argon?
Answer: This typically indicates hydrocarbon contamination from either argon gas impurities or improper sample handling.
Troubleshooting Steps:
- Check Argon Gas Purity: Verify argon purity is 99.999% or higher for surface analysis
- Inspect Gas Delivery System: Examine regulators, valves, and tubing for leaks or contamination
- Validate Sample Handling Protocol: Ensure gloves and tools are contamination-free
- Test Chamber Base Pressure: Confirm analysis chamber maintains < 5 × 10⁻⁹ mbar
Quantitative Analysis of Common Contaminants:
| Contaminant Source | Typical Concentration | Impact on XPS Signal | Detection Method |
|---|---|---|---|
| Argon (Impure) | 10-100 ppm hydrocarbons | Increased C 1s, O 1s | RGA, Blank spectra |
| Fingerprints | ~10¹⁵ carbon atoms/cm² | Dominant C 1s peak | Visual inspection |
| Pump Oil | Varies by system age | Hydrocarbon envelope | RGA monitoring |
| Chamber Outgassing | 10⁻⁸-10⁻⁷ mbar | Gradual contamination | Pressure rise rate |
Problem: Inconsistent Spectroscopy Results
Question: Why do I get varying results between replicate samples prepared under "identical" conditions?
Answer: Inconsistent handling introduces variable contamination that affects spectroscopic measurements.
Diagnostic Protocol:
Frequently Asked Questions
Argon-Related Issues
Q1: What argon purity level is sufficient for sensitive spectroscopy applications? A: For surface-sensitive techniques like XPS or ToF-SIMS, use 99.999% (5.0 grade) or higher purity argon. Lower grades contain hydrocarbons and moisture that deposit on samples.
Q2: How can I verify argon quality in my laboratory? A: Implement these verification methods:
- Residual Gas Analysis (RGA) of sputtering chamber
- Blank runs with inert reference samples
- Regular gas certification from suppliers
Q3: What are the signs of argon system contamination? A: Key indicators include:
- Gradual increase in carbon background over time
- Unusual peaks in mass spectrometry
- Reduced sputtering rates
- Inconsistent sample charging
Sample Handling Issues
Q4: What is the proper glove protocol for sensitive sample preparation? A: Follow this optimized procedure:
Q5: How does improper handling affect drug development research? A: Contamination introduces significant errors in:
- Surface composition analysis of drug formulations
- Catalyst characterization in synthesis pathways
- Biomaterial interface studies
- Quality control measurements
Experimental Protocols
Protocol 1: Argon Purity Verification Method
Objective: Quantify hydrocarbon contaminants in argon supply
Materials:
- Residual Gas Analyzer (RGA)
- High-vacuum chamber
- Reference sample (clean silicon wafer)
- Gas purification filter (optional)
Procedure:
- Evacuate chamber to base pressure < 1 × 10⁻⁸ mbar
- Record RGA background spectrum
- Introduce argon gas at working pressure (typically 5 × 10⁻³ mbar)
- Acquire RGA spectrum focusing on mass 12-16 (carbon), 18 (water), 28 (CO/N₂)
- Compare contaminant peaks against calibration standards
Acceptance Criteria:
| Contaminant | Maximum Allowable Level | Typical Mass Peak |
|---|---|---|
| Hydrocarbons | < 1 ppm | 15, 27, 29, 41, 43 |
| Water | < 3 ppm | 18 |
| Oxygen | < 2 ppm | 32 |
| Nitrogen | < 5 ppm | 14, 28 |
Protocol 2: Contamination-Free Sample Transfer
Objective: Transfer samples from preparation chamber to analysis position without introducing contaminants
Workflow:
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function | Critical Specifications |
|---|---|---|
| Ultra-High Purity Argon | Sputtering gas and atmosphere control | 99.999% purity, hydrocarbon < 0.5 ppm |
| Powder-Free Nitrile Gloves | Sample handling protection | Low extractables, sulfur and chloride free |
| High-Purity Isopropanol | Surface cleaning solvent | ≥99.9%, filtered through 0.2 μm membrane |
| Ceramic Tweezers | Sample manipulation | Non-magnetic, anti-static coating |
| Stainless Steel Transfer Rods | Inter-chamber sample movement | Magnetically coupled, bakeable to 150°C |
| RGA System | Gas quality monitoring | Mass range 1-100 amu, detection limit < 10⁻¹² mbar |
| Vacuum-Compatible Samples Holders | Sample mounting | Machinable materials (Ta, Mo, stainless steel) |
| In-Situ Sample Cleaver | Creating fresh surfaces | UHV compatible, impact energy controlled |
This guide provides troubleshooting procedures for common hardware issues affecting spectrometer data quality. Proper maintenance of lens alignment and probe contact is critical for acquiring reliable spectral data for your research.
Frequently Asked Questions
1. Why is lens alignment critical in spectroscopy? The lens must focus precisely on the light source to collect an adequate amount of light for measurement. Improper alignment means the instrument collects less light, leading to low-intensity spectra and highly inaccurate quantitative results [75].
2. What are the symptoms of incorrect spectrometer probe contact? You may encounter a louder-than-usual sound during metal analysis and see a bright light escaping from the pistol face. This often results in incorrect results or a complete failure to acquire data. Severe cases can cause high voltage to discharge inside the connector, which is dangerous and costly to repair [75].
3. How often should I perform these maintenance checks? As a manufacturer-recommended best practice, a full scheduled maintenance visit, including checks of optical systems and mechanical components, should be performed at least once per year. Instruments used extensively or in demanding environments may require more frequent checks [76] [77].
Troubleshooting Guide: Symptoms and Solutions
The table below summarizes common problems, their symptoms, and immediate corrective actions.
Table 1: Troubleshooting Common Lens and Probe Issues
| Component | Observed Symptom | Potential Cause | Corrective Action |
|---|---|---|---|
| Lens Alignment | Consistently low intensity spectra; inaccurate readings for all elements [75]. | Lens is misaligned and not focused on the light source [75]. | Perform lens alignment procedure as part of regular operator maintenance; replace lens if damaged [75]. |
| Probe Contact | Loud analysis sound; bright light from pistol face; no results or poor results [75]. | Poor contact with the sample surface; irregular sample shape [75]. | Increase argon flow from 43 psi to 60 psi; use seals for convex shapes; consult a technician for a custom-built pistol head [75]. |
| Optical Windows | Instrument analysis drifts frequently, requiring more recalibration; poor analysis reading [75]. | Dirty windows in front of the fiber optic cable or in the direct light pipe [75]. | Clean the optical windows regularly as part of a scheduled maintenance routine [75]. |
Detailed Experimental Protocols
Protocol 1: Verifying and Correcting Lens Alignment
Improper lens alignment is a common issue that leads to a loss of light intensity and erroneous results. The following procedure outlines the corrective actions.
Table 2: Essential Materials for Lens Alignment
| Item | Function |
|---|---|
| High-Precision Alignment System (e.g., OptiCentric) | Provides precise measurement and alignment of lenses with accuracy down to 0.1 µm [78]. |
| Calibrated Reference Sample | A standard sample with a known spectral signature to verify the accuracy of the realignment. |
| Manufacturer's Software | Software tools (e.g., MultiLens, SmartAlign) are used for quality checks and aligning the optical axis [78]. |
Methodology:
- Operator Checks: Train operators to perform simple lens alignment fixes and recognize when a lens needs replacement. This is typically an easy task [75].
- High-Precision Alignment: For critical applications or complex lens assemblies, use a dedicated system like an OptiCentric device.
- Mount the lens or assembly on the motorized stage.
- Use the system's software to measure the centration error (the deviation of the optical axis from the mechanical axis).
- The software provides guidance for manual or automated realignment. The patented SmartAlign module, for instance, can use the optical axis itself as a reference for alignment [78].
- Verify alignment by measuring the reference sample and confirming the spectral output matches expected intensity and values.
Protocol 2: Troubleshooting Probe Contact Issues
Incorrect probe contact prevents proper sample excitation and compromises data integrity. This protocol addresses this critical interface.
Table 3: Essential Materials for Probe Contact Troubleshooting
| Item | Function |
|---|---|
| Argon Gas Supply & Flow Regulator | Provides and controls the argon gas flow (typically increased to 60 psi) to improve the analysis environment [75]. |
| Convex Seals | Specialized seals that help create a flush contact between the probe and a curved sample surface [75]. |
| Custom Pistol Head | A technician-built probe head designed to accommodate highly irregular surface contours [75]. |
Methodology:
- Increase Argon Flow: If you observe symptoms of poor contact, increase the argon flow from a typical 43 psi to 60 psi. This can help stabilize the analysis spot [75].
- Use Specialized Seals: For samples with convex or curved surfaces, add appropriate seals to the probe face to create a better seal [75].
- Custom Solutions: For consistently irregularly shaped samples, contact a technical service provider to custom-build a pistol head that matches the specific contours of your samples [75].
- Sample Preparation: Ensure samples are properly prepared. They should be ground flat and not contaminated by oils from skin contact, which can also affect analysis [75].
Workflow for Diagnosing Hardware Issues
The following diagram illustrates a logical workflow for diagnosing and addressing the hardware issues discussed in this guide.
Troubleshooting Guides
FAQ: Addressing Common Automated Liquid Handling and HTS Challenges
1. My HTS data is inconsistent between runs. How can I identify if the liquid handler is the source? Inconsistent data often stems from liquid handling errors. To troubleshoot, first verify that the liquid handler is functioning correctly. Key factors to check include the instrument's calibration, the properties of the liquids being dispensed (e.g., viscosity, volatility), and the environmental conditions. Automated systems with built-in verification features, such as DropDetection technology, can proactively identify and document dispensing errors, which is the first step in resolving variability issues [79].
2. What are the best practices for ensuring data reproducibility in automated workflows? Ensuring reproducibility requires a multi-pronged approach focusing on standardization and error reduction [80] [79].
- Standardize Protocols: Automated workflows significantly reduce inter- and intra-user variability by performing liquid handling, plate management, and reagent dispensing identically every time [80].
- Implement In-process Verification: Utilize technologies that confirm successful liquid transfers in real-time. This allows for the immediate flagging of errors, preventing unreliable data from progressing through the screening pipeline [79].
- Automate Data Management: Integrate software solutions that automatically collect and process the vast amounts of data generated by HTS. This reduces manual, error-prone data entry and analysis, leading to more reliable and reproducible insights [80] [79].
3. How can I reduce the high costs associated with HTS reagent consumption? Automation enables miniaturization, which is key to cost reduction. By using non-contact dispensers that can accurately handle volumes in the nanoliter range (as low as 4 nL), you can scale down reaction volumes dramatically. This approach can reduce reagent consumption and associated costs by up to 90% while maintaining or even improving data quality [80] [79].
4. My lab is new to automation. What should we consider when implementing an automated HTS workflow? Successful implementation begins with a careful assessment of your current processes [79].
- Identify Bottlenecks: Pinpoint the manual, labor-intensive tasks in your workflow, such as serial dilutions or multi-step assay preparations, that would benefit most from automation.
- Select Flexible Tools: Choose scalable automation tools that can adapt to your project's needs. For liquid handling, consider platforms that offer high precision at low volumes and can be integrated into larger automated work cells for future expansion [79].
- Evaluate Support: Ensure the technology provider offers robust technical support, user-friendly software, and training to guarantee a smooth transition and sustained operation [79].
Table 1: Impact of Automation on High-Throughput Screening (HTS)
| Metric | Impact of Automation | Key Benefit |
|---|---|---|
| Throughput | Enables screening of up to 50,000 wells per day [81] | Accelerated lead compound identification |
| Reagent Cost | Reduction of up to 90% through miniaturization [79] | More sustainable and cost-effective research |
| Data Reproducibility | Reduces human error and inter-user variability [80] [79] | Increased reliability of screening results |
| Liquid Handling Precision | Non-contact dispensing as low as 4 nL [80] | Enables miniaturization and saves precious reagents |
Table 2: Spectroscopy Software Market and Application Trends
| Category | Detail | Significance |
|---|---|---|
| Global Market Size (2024) | ~USD 1.1 Billion [1] | Demonstrates widespread adoption in analytical labs |
| Projected CAGR (2025-2034) | 9.1% [1] | Indicates strong and sustained growth |
| Pharmaceutical Segment Share (2024) | 28.9% [1] | Highlights its critical role in drug discovery and quality control |
| Key Software Trend | Integration of AI and ML for data analysis [1] | Enables faster data processing, pattern detection, and predictive analytics |
Experimental Protocol: Automated Flow Cytometry for Phenotypic Screening
This protocol outlines a fully automated high-throughput flow cytometry screening workflow for complex phenotypic assays [81].
1. Principle Phenotypic screening allows for drug discovery without prior knowledge of a specific molecular target by using primary cells or co-culture models that closely mimic the disease pathology. Automated flow cytometry enables multiparametric, single-cell analysis of these complex models at high throughput.
2. Materials
- Cells: Primary human cells (e.g., CD4+ T cells, CD34+ hematopoietic stem cells) or cell lines [81].
- Liquid Handler: Automated system capable of dispensing into 384-well plates [81].
- Flow Cytometer: High-throughput capable instrument (e.g., CyAn ADP) [81].
- Assay Reagents: Cell culture media, stimulation beads (e.g., anti-CD3/anti-CD28), cytokines (e.g., TGF-β), staining antibodies, fixation/permeabilization buffers [81].
3. Procedure
- Step 1: Cell Preparation and Plating
- Isolate primary cells using immunoselection kits according to manufacturer instructions.
- Resuspend cells in culture media at a concentration of 0.5 x 10^6 cells/mL.
- Using an automated liquid handler, dispense cells into 384-well plates pre-dispensed with compounds or controls [81].
- Step 2: Cell Stimulation and Culture
- Step 3: Automated Staining
- Perform surface staining for markers (e.g., CD4, CD25) using an automated online system.
- For intracellular targets (e.g., Foxp3), perform fixation and permeabilization using commercial buffer sets, followed by staining with the target antibody—all via the automated workflow [81].
- Step 4: Data Acquisition and Analysis
- Acquire samples directly from the 384-well plates using the high-throughput flow cytometer.
- Use automated data analysis software to process the multiparametric data and identify hit compounds based on the desired phenotypic signature [81].
Workflow Visualization
Automated HTS Troubleshooting Logic
Raman Spectroscopy with MVDA Workflow
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Reagents and Materials for Automated HTS and Spectroscopy
| Item | Function | Application Example |
|---|---|---|
| I.DOT Liquid Handler | Non-contact dispensing with low volume (4 nL) precision and DropDetection verification [80] [79] | Accurate compound and reagent dispensing in HTS assays |
| Multivariate Data Analysis (MVDA) Software (e.g., SIMCA) | Finds correlations between spectral data and reference analytics to build predictive calibration models [72] | Analyzing Raman spectroscopy data for bioprocess monitoring |
| Design of Experiments (DOE) Software (e.g., MODDE) | Statistical approach to design efficient experiments for building robust models with fewer runs [72] | Planning Raman calibration experiments with optimal parameter variations |
| Fluorescent Barcoding Kit (e.g., FluoReporter) | Labels different cell populations with unique fluorescent tags for multiplexed analysis [81] | Screening hybridoma supernatants against multiple cell lines in a single well |
| Process Analytical Technology (PAT) Tools (e.g., Raman Spectrometer) | Integrated, non-invasive sensors for real-time monitoring of critical process parameters [72] | Monitoring glucose, lactate, and other metabolites in a bioreactor |
Validation and Comparative Analysis: Ensuring Accuracy and Selecting the Right Tools
Core Principles and Key Parameters of Method Validation
Analytical method validation is a critical process that provides definitive evidence that an analytical procedure is suitable for its intended purpose, ensuring the reliability, accuracy, and reproducibility of results, which is paramount in fields like pharmaceutical development [82] [83].
The table below summarizes the key parameters evaluated during method validation and their core function in ensuring data quality.
Table 1: Key Analytical Method Validation Parameters
| Parameter | Description | Purpose in Ensuring Quality |
|---|---|---|
| Accuracy [84] [85] | Closeness of agreement between measured value and true value. | Demonstrates the method yields truthful results, often assessed by spiking known amounts of analyte. |
| Precision [84] [85] | Closeness of agreement between a series of measurements. Includes repeatability (intra-day) and reproducibility (inter-day). | Ensures the method produces consistent results under prescribed conditions. |
| Specificity/Selectivity [86] [84] [85] | Ability to measure the analyte accurately in the presence of potential interferences. | Confirms the method can distinguish and quantify the target analyte from other components. |
| Linearity [84] [85] | Ability to obtain results directly proportional to analyte concentration. | Verifies the method's proportional response across a defined range. |
| Range [85] | The interval between upper and lower concentrations with suitable precision, accuracy, and linearity. | Defines the concentrations over which the method is applicable. |
| Limit of Detection (LOD) [82] [85] | Lowest concentration of analyte that can be detected. | Establishes the method's sensitivity for detecting trace amounts. |
| Limit of Quantitation (LOQ) [82] [85] | Lowest concentration of analyte that can be quantified with acceptable precision and accuracy. | Establishes the method's sensitivity for reliable quantification. |
| Robustness [86] [85] | Capacity to remain unaffected by small, deliberate variations in method parameters. | Indicates the method's reliability during normal use and its susceptibility to minor changes. |
Troubleshooting Common Method Validation & Instrument Issues
Method Validation Troubleshooting Guide
Table 2: Common Method Validation Issues and Solutions
| Problem | Possible Cause | Recommended Solution |
|---|---|---|
| Method is not robust [86] | Investigating robustness for the first time during formal validation. | Investigate robustness during method development using a specific protocol, before validation begins [86]. |
| Investigating wrong robustness factors [86] | Focusing only on instrument parameters while ignoring sample preparation. | Use a Subject Matter Expert (SME) to review all method steps, especially those adjusted during development [86]. |
| Poor Specificity [83] | Interference from sample matrix, impurities, or degradation products. | Evaluate potential interferences during validation. Techniques like chromatographic separation can help ascertain specificity [84] [83]. |
| Failing Regulatory Audit [83] | Incomplete reporting of validation data; only reporting results within acceptable limits. | Report all validation results, both passing and failing, to provide a complete data picture for regulators [83]. |
Spectrometer Troubleshooting Guide
Table 3: Common Spectrometer Issues and Solutions
| Problem | Possible Cause | Recommended Solution |
|---|---|---|
| Unstable/Drifting Readings [87] | Instrument lamp not warmed up; air bubbles in sample; sample too concentrated. | Allow 15-30 minutes for lamp warm-up; gently tap cuvette to dislodge bubbles; dilute sample [87]. |
| Inaccurate Analysis Results [75] | Loss of intensity for low-wavelength elements (e.g., Carbon, Phosphorus). | Check the vacuum pump; monitor for constant low readings for carbon, phosphorus, and sulfur [75]. |
| Cannot Set to 100% Transmittance (Fails to Blank) [87] | Light source (lamp) is near end of life; dirty optics. | Check lamp usage hours; replace if old. If optics are dirty, seek professional servicing [87]. |
| Negative Absorbance Readings [87] | The blank solution was "dirtier" than the sample; different cuvettes used for blank and sample. | Use the exact same cuvette for both blank and sample measurements; ensure cuvettes are clean [87]. |
| Contaminated Argon [75] | Contaminated argon supply or sample. | Regrind samples with a new grinding pad; ensure samples are not quenched in water/oil or touched with bare hands [75]. |
Frequently Asked Questions (FAQs)
Q1: What is the difference between LOD and LOQ? The Limit of Detection (LOD) is the lowest concentration at which the analyte can be reliably detected, but not necessarily quantified. The Limit of Quantitation (LOQ) is the lowest concentration that can be quantified with acceptable precision and accuracy [82] [85].
Q2: When should robustness be evaluated? Robustness should be investigated during the method development phase, not during formal validation. This ensures any issues are resolved before the final method is locked and full validation begins, preventing invalidated results [86].
Q3: How can I ensure my method is compliant with regulatory requirements? To ensure compliance, develop a formal validation plan outlining the protocol and acceptance criteria, use validated software for data analysis, and maintain detailed documentation of all activities and results [82] [83]. Key guidelines include ICH Q2(R1) and FDA Guidance for Industry on Bioanalytical Method Validation [82].
Q4: What are common pitfalls in interpreting validation data? Common pitfalls include collecting insufficient data for reliable conclusions, using incorrect statistical methods, and failing to account for variability, which can lead to an overestimation of method performance [82].
Q5: The software for my spectrometer is complex. What trends are making it easier to use? The market is seeing key developers invest in creating more user-friendly dashboards for non-specialists. Modern software packages increasingly include intuitive interfaces, automated workflows, and customizable reporting to improve accessibility [1].
Experimental Workflow for Method Validation
The following diagram outlines a generalized workflow for the analytical method validation process, from defining requirements to final validation, incorporating principles of good experimental design.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 4: Essential Materials for Spectroscopy and Analytical Methods
| Item | Function |
|---|---|
| Certified Reference Materials (CRMs) [82] | Used to validate method accuracy by providing a sample with a known, certified value for the analyte. |
| High-Purity Solvents & Reagents [83] | Essential for sample preparation, mobile phases (in chromatography), and blank solutions to prevent interference and baseline noise. |
| Ultrapure Water [7] | Critical for sample preparation, dilution, and preparation of buffers and mobile phases, ensuring no contaminants affect the analysis. |
| Quartz Cuvettes [87] | Required for measurements in the ultraviolet (UV) range, as they allow UV light to pass through, unlike plastic or glass. |
| Stable, Homogeneous Samples [82] | The foundation of any valid analysis. Samples must be properly prepared, homogeneous, and stable during storage and analysis. |
| Chromatography Columns (HPLC/GC) [86] | A key consumable in chromatographic methods. Batch-to-batch variation in columns is a common factor tested in robustness studies. |
Frequently Asked Questions (FAQs)
Q1: What is the fundamental difference between LOD and LOQ?
The Limit of Detection (LOD) is the lowest concentration of an analyte that can be reliably distinguished from a blank sample, but not necessarily quantified with precise accuracy. In contrast, the Limit of Quantitation (LOQ) is the lowest concentration at which the analyte can not only be detected but also quantified with acceptable precision and accuracy, meeting predefined goals for bias and imprecision [88] [89]. The LOQ is always greater than or equal to the LOD [89].
Q2: Why is there a risk of false negatives at the LOD?
At the LOD concentration, there is a significant overlap between the distribution of measurement signals from a blank sample and the distribution of signals from a sample containing the analyte at the LOD. This overlap means that a sample containing the analyte at the LOD has a substantial probability (typically set at 5% or 50%, depending on the definition used) of producing a signal below the critical decision level, leading to the false conclusion that the analyte is not present (a false negative) [90].
Q3: How do Instrument Detection Limit (ILD) and Method Detection Limit (MDL) differ?
The Instrument Detection Limit (ILD or IDL) is the minimum net signal or concentration detectable by the analytical instrument itself, typically defined with a high confidence level (e.g., 99.95%) [91]. The Method Detection Limit (MDL) is a "global" detection limit that includes all steps of the analytical method, such as sample preparation, digestion, dilution, or concentration. These additional steps introduce more opportunities for error, making the MDL higher than the IDL [88].
Q4: What are the common methods for determining LOD and LOQ?
Common approaches include the statistical method, the signal-to-noise ratio method, and visual inspection [92]. The statistical method uses the standard deviation of the response and the slope of the calibration curve: LOD = 3.3(SD/S) and LOQ = 10(SD/S), where SD is the standard deviation and S is the slope [92]. The signal-to-noise ratio method defines the LOD as a concentration that yields a signal typically 3 to 3.3 times the noise level, while the LOQ is typically 10 times the noise [90] [91].
Q5: How do modern spectroscopy software tools assist with detection limit calculations?
Modern spectroscopy software is increasingly incorporating artificial intelligence (AI) and machine learning (ML) to improve data gathering, analysis, and interpretation [1]. These tools enable better and faster processing of spectral data, pattern detection, and predictive analytics. Many software platforms also offer automated workflows and customizable reporting, which can streamline the process of determining and validating detection limits [1] [7].
Troubleshooting Guides
Issue 1: Unacceptably High or Variable Detection Limits
Potential Causes and Solutions:
- Cause: High Background Noise. Excessive or fluctuating background noise can mask the analyte signal, raising the apparent detection limit.
- Solution: Ensure instrument optics and source are clean. Use high-purity reagents and solvents to minimize chemical background. Verify that the instrument is properly calibrated and maintained. Use spectral filtering or software-based background correction techniques if supported by your system [88] [91].
- Cause: Poor Analytical Sensitivity. A low response (slope of the calibration curve) for a given concentration change makes it harder to distinguish the analyte from the blank.
- Cause: Sample Matrix Effects. The sample matrix can suppress or enhance the analyte signal, or contribute to the background, directly impacting the standard deviation of the measurement.
- Solution: Employ sample preparation techniques to clean up and concentrate the sample, such as solid-phase extraction. Use the method of standard additions to correct for matrix effects. Ensure that the blank matrix used for LOD/LOQ determination is commutable with the real sample matrix [89].
Issue 2: Inconsistent LOD/LOQ Values When Using Different Calculation Methods
Potential Causes and Solutions:
- Cause: Non-Linear Response at Low Concentrations. The statistical approach (using the calibration curve slope) assumes linearity. If the response is non-linear near the detection limit, the calculated values will be unreliable.
- Solution: Visually inspect the calibration plot at low concentrations. If non-linearity is observed, use a different calculation method, such as signal-to-noise, or prepare additional low-level standards to better define the curve's behavior in that region [92].
- Cause: Insufficient or Low-Quality Replicate Data. The standard deviation used in calculations is highly sensitive to the number and quality of replicate measurements.
- Solution: Follow established protocols that recommend a sufficient number of replicates (e.g., CLSI EP17 recommends 60 for establishment and 20 for verification by a manufacturer) [89]. Ensure that replicate preparations and measurements are performed independently to capture all sources of method variability.
Issue 3: Reported Results Near the LOD Show High Imprecision
Potential Causes and Solutions:
- Cause: Reporting Results Below the LOQ. The LOD is a limit of detection, not a limit of reliable quantification. Reporting numerical results at or near the LOD will naturally have high imprecision.
- Cause: Inappropriate Data Reporting Intervals. The reporting interval (the number of decimal places) may be finer than the method's precision can support at low concentrations.
- Solution: Align the reporting interval with the method's precision. A common rule (ASTM E-29-02) is that results should be rounded to no less than 1/20 of the determined standard deviation. For example, a method with a standard deviation of 1.1 should have a reporting interval of 0.1 [92].
Experimental Protocols
Protocol 1: Estimating LOD and LOQ using the Statistical and Calibration Curve Method
This is a commonly practiced and robust approach recommended by guidelines such as ICH Q2(R1) [92].
Methodology:
- Preparation of Standards: Prepare a series of standard solutions at a minimum of five concentration levels, with the lowest levels in the region where the detection limit is expected. The standards should be prepared in the same matrix as the sample.
- Analysis: Analyze each standard solution in replicate (e.g., 3-5 times each) following the complete analytical procedure.
- Calibration Curve: Plot the average measured response (e.g., peak area, absorbance) against the known concentration for each standard.
- Linear Regression: Perform a linear regression analysis on the data to obtain the slope (S) and the standard deviation of the response (SD). The SD can often be derived as the standard error of the y-intercept or from the residuals of the regression [92].
- Calculation:
Protocol 2: Empirical Determination of LOD and LOQ using Blank and Low-Level Samples
This method, aligned with CLSI EP17, provides an empirical estimate that captures the variability of the actual sample matrix [89].
Methodology:
- Sample Preparation:
- Blank Sample: Obtain or prepare a sample that is identical to the test sample but contains no analyte.
- Low-Concentration Sample: Prepare a sample containing the analyte at a concentration near the expected LOD.
- Replicate Analysis: Analyze a minimum of 20 replicates of both the blank sample and the low-concentration sample. For a full validation, 60 replicates are recommended [89].
- Data Analysis:
- Calculate the mean (
mean_blank) and standard deviation (SD_blank) of the results from the blank. - Calculate the mean and standard deviation (
SD_low) of the results from the low-concentration sample.
- Calculate the mean (
- Calculation:
Data Presentation
| Term | Acronym | Definition | Key Feature |
|---|---|---|---|
| Limit of Blank | LoB | The highest apparent analyte concentration expected to be found when replicates of a blank sample (containing no analyte) are tested [89]. | Measures the background noise of the method. |
| Limit of Detection | LOD / LoD | The lowest analyte concentration that can be reliably distinguished from the LoB. It is the level at which detection is feasible, but not necessarily quantifiable with acceptable precision [88] [89]. | Associated with a low probability of false negatives. |
| Instrument Detection Limit | IDL / ILD | The minimum net signal or concentration of an analyte that can be detected by the instrument alone in a given analytical context, often with a 99.95% confidence level [91]. | Specific to the instrument's performance, excluding sample preparation. |
| Method Detection Limit | MDL | The lowest concentration of an analyte that can be detected after undergoing the complete analytical method, including sample preparation [88]. | A "global" detection limit that is always higher than the IDL. |
| Limit of Quantitation | LOQ / LoQ | The lowest concentration of an analyte that can be quantified with acceptable precision (bias and imprecision) [88] [89]. | Defines the lower limit of the quantitative range of an assay. |
Table 2: Comparison of Common LOD/LOQ Determination Methods
| Method | Description | Typical Formula (for LOD) | Advantages | Limitations |
|---|---|---|---|---|
| Statistical (Calibration Curve) | Uses the standard deviation of the response and the slope of the calibration curve [92]. | 3.3 * (SD / S) |
Objective; uses data from the actual calibration; widely accepted. | Assumes linearity at low concentrations; SD can be difficult to estimate accurately. |
| Signal-to-Noise | Directly compares the magnitude of the analyte signal to the background noise [90] [91]. | Signal = 3 × Noise | Simple, intuitive, and instrument-independent; commonly used in chromatography. | Can be subjective in measuring noise; may not capture all sources of method variability. |
| Empirical (EP17) | Based on the statistical analysis of replicate measurements of a blank and a low-concentration sample [89]. | LoD = LoB + 1.645 * SD_low |
Empirically measures performance in the sample matrix; robust and statistically sound. | Requires a large number of replicate measurements; more labor-intensive. |
Visualizations
Detection Limit Relationship Diagram
LOD/LOQ Experimental Workflow
The Scientist's Toolkit
Table 3: Essential Research Reagent and Material Solutions
| Item | Function in Detection Limit Analysis |
|---|---|
| High-Purity Blank Matrix | A sample material identical to the test sample but devoid of the target analyte. Used to determine the LoB and the baseline noise of the method [89]. |
| Certified Reference Materials (CRMs) | Standards with known, traceable concentrations of the analyte. Essential for preparing accurate calibration standards and low-concentration samples for empirical LoD determination [91]. |
| High-Purity Solvents and Reagents | Minimize chemical background noise and interference in the spectral or chromatographic baseline, which is critical for achieving low detection limits [88]. |
| Stable, Low-Concentration QC Sample | A quality control sample with an analyte concentration near the expected LOD or LOQ. Used to continuously verify the method's detection capability over time [89]. |
| Appropriative Spectroscopy Software | Software with tools for signal processing, noise calculation, statistical analysis (including regression), and automated reporting. Modern software often includes AI/ML features for enhanced data analysis [1] [7]. |
X-ray Fluorescence (XRF) spectrometry is a powerful, non-destructive analytical technique used to determine the elemental composition of materials, making it indispensable for the analysis of complex alloy systems [93]. When an atom is irradiated with high-energy X-rays, inner-shell electrons are ejected. As electrons from outer shells fall to fill these vacancies, they emit fluorescent X-rays with energies characteristic of the element from which they originated [94] [93]. This fundamental process allows for both qualitative and quantitative analysis of solid samples, including a wide range of metal alloys [93].
There are two primary configurations of XRF spectrometers: Energy Dispersive XRF (ED-XRF) and Wavelength Dispersive XRF (WD-XRF) [95] [94]. The core difference between them lies in how they detect and measure the emitted X-rays. ED-XRF uses a semiconductor detector to simultaneously measure the energies of the incoming X-ray photons, converting them into an electrical signal to generate a complete fluorescence energy spectrum [95] [96] [94]. In contrast, WD-XRF employs an analyzing crystal to disperse the fluorescent X-rays according to their wavelengths (based on Bragg's Law), and a detector measures the intensity of each wavelength sequentially [95] [94]. This fundamental distinction in detection philosophy leads to significant differences in performance, applicability, and operational requirements, which are critical to understand when selecting the optimal technique for analyzing complex alloys.
Technical Comparison: ED-XRF vs. WD-XRF
The choice between ED-XRF and WD-XRF involves balancing multiple factors, including analytical performance, operational speed, and cost. The following table summarizes the core technical differences relevant to alloy analysis.
Table 1: Technical Comparison of ED-XRF and WD-XRF for Alloy Analysis
| Feature | ED-XRF | WD-XRF |
|---|---|---|
| Detection Principle | Measures X-ray energies directly with a solid-state detector [94] | Disperses X-rays by wavelength using a diffraction crystal [95] [94] |
| Analysis Speed | Very fast; simultaneous multi-element detection (seconds per sample) [97] | Slower; sequential or fixed-channel measurement (minutes per sample) [98] [97] |
| Spectral Resolution | Lower (typically 150 eV - 300 eV) [94] | Higher (typically 5 eV - 20 eV) [94] |
| Typical Detection Limits | ppm to % levels [97] | ppb to ppm levels [97] |
| Light Element Analysis (e.g., Be, C) | Limited capability; typically effective from Sodium (Na) and heavier [94] [97] | Excellent capability; can analyze down to Beryllium (Be) [94] [97] |
| Portability | High; handheld and portable benchtop models available [97] [93] | Low; typically large, lab-bound systems [97] [93] |
| Initial and Operational Cost | Generally lower [98] [97] | Significantly higher investment and maintenance [98] [97] |
| Ease of Use | Simple operation, minimal training required [96] | More complex, requires specialized operators [93] |
For alloy analysis, these technical differences translate directly into practical advantages and limitations. The higher resolution and superior detection limits of WD-XRF make it the definitive choice for quantifying trace elements and accurately measuring major concentrations in complex matrices with high precision [98] [93]. Its ability to analyze light elements like carbon and boron is also critical for certain alloy systems [97]. Conversely, the speed, portability, and lower cost of ED-XRF make it ideal for rapid alloy identification, material sorting, and on-site verification [97]. Its simultaneous detection capability provides a quick elemental overview, which is often sufficient for quality control and grade verification purposes.
Troubleshooting Guides and FAQs
Common Operational Issues and Solutions
Users of XRF spectrometers often encounter operational issues that can affect data quality. Below is a troubleshooting guide for common problems.
Table 2: XRF Troubleshooting Guide for Common Operational Issues
| Problem | Potential Causes | Corrective Actions |
|---|---|---|
| Low Count Rates/Instrument will not start analysis | Sample not properly presented in front of the window; contaminated or damaged detector window; depleted battery (for handheld units) [99] | Ensure sample completely covers the measurement window. Clean or replace the detector window. Check and charge or replace the battery [99]. |
| Poor Precision (High variability between measurements) | Insufficient measurement time; sample heterogeneity; loose or vibrating instrument components [100] | Increase counting time to improve counting statistics. For heterogeneous materials like alloys, take 3-5 readings at different spots [100]. Ensure the instrument is on a stable surface. |
| Inaccurate Results vs. Certified Reference Materials | Incorrect or outdated calibration; spectral interferences; sample surface effects (e.g., oxidation, roughness) [100] [93] | Recalibrate the instrument with certified standard materials. Use software tools to correct for spectral overlaps. Clean, polish, or re-machine the sample surface to ensure a flat, representative analysis area [100]. |
| Unusual Spectral Peaks or High Background | Contamination of the sample cup or instrument window; instrument malfunction or detector drift [99] [100] | Run a blank (e.g., silica blank) to check for contamination. Clean or replace the sample cup and window. Perform an energy calibration check using a standard (e.g., SS316); if it fails, contact service [99]. |
| Software Errors or Instrument Freezing | Software glitch; operating system conflict [99] | A simple restart often resolves minor software errors. Turn the instrument off and back on [99]. |
Frequently Asked Questions (FAQs)
Q1: Our lab needs to analyze stainless steel for major constituents (Cr, Ni, Fe) and also quantify trace-level tramp elements (e.g., Pb, Sn). Which technique is more suitable? For this dual requirement, WD-XRF is the superior choice for laboratory-based analysis. Its high resolution allows for the precise separation and accurate quantification of major elements like Cr and Ni, which have closely spaced spectral lines. Furthermore, its lower detection limits are essential for reliably measuring trace tramp elements at low ppm concentrations [98] [93]. While ED-XRF can screen for these elements, WD-XRF provides the analytical rigor needed for certification and high-precision quality control.
Q2: We need to sort hundreds of scrap metal pieces in a yard quickly. Is WD-XRF a viable option? No, for this application, handheld ED-XRF is the definitive solution. The portability of handheld ED-XRF allows you to take the instrument to the scrap piles. Its analysis speed of a few seconds per piece enables rapid sorting of alloys like distinguishing between 304 and 316 stainless steel [97]. The non-destructive nature also preserves the value of the scrap. WD-XRF is not portable and is far too slow for high-throughput sorting, making it impractical for this task [97].
Q3: Why are our results for a copper-tin alloy inconsistent, even though the sample appears homogeneous? This is a classic sign of sample heterogeneity at the microscopic level. Even if an alloy appears uniform, elements can segregate during solidification, creating micro-inhomogeneities [100]. The small analysis spot of an XRF spectrometer may be reading different micro-constituencies. To mitigate this, ensure you are using a representative sample and take multiple readings (3-5) from different locations on the sample and average the results. Also, verify that the sample surface is clean, flat, and properly prepared [100].
Q4: What is the most critical step to ensure accurate quantitative analysis of a new alloy type? The single most critical step is proper calibration using certified reference materials (CRMs) that closely match the new alloy's composition and matrix [100] [93]. Using an incorrect calibration (e.g., a pure metal standard for a complex alloy) will lead to significant inaccuracies due to matrix effects, where elements influence each other's X-ray intensities. Always use matrix-matched standards for the highest accuracy [93].
Experimental Protocols for Alloy Analysis
Sample Preparation Workflow
Proper sample preparation is paramount for achieving accurate and reproducible results in XRF analysis. Inconsistent preparation is a leading cause of analytical error [100]. The following diagram outlines a standard workflow for preparing solid alloy samples.
Sample Preparation Workflow
Key Steps Explained:
- Visual Inspection and Cleaning: Examine the sample for surface contamination, heavy corrosion, or coatings. Clean the surface with a solvent like acetone or ethanol to remove oils and dirt [100].
- Decision Point: The choice between non-destructive and destructive preparation is critical. Non-destructive analysis is used for finished products, valuable components, or when the sample must be preserved. The sample is analyzed as-is, but the surface should be as flat as possible for optimal results [93].
- Destructive Preparation (for highest accuracy):
- Cutting: Obtain a representative section of the alloy using a saw or cutter. Avoid overheating, which can alter the microstructure.
- Machining/Polishing: This is the most important step for quantitative analysis. Create a flat, smooth surface by turning on a lathe or polishing with abrasive papers (e.g., SiC paper down to 400-600 grit). A smooth surface minimizes topography effects and improves reproducibility [100] [93].
- Final Cleaning: Remove any metal dust or polishing debris from the newly created surface before analysis.
Technique Selection Protocol
Selecting the appropriate XRF technique is a strategic decision based on analytical requirements and operational constraints. The following decision tree guides users through this process.
Technique Selection Protocol
Instrument Calibration and Verification Procedure
Objective: To establish and verify a calibration curve for quantitative analysis of a specific alloy type (e.g., stainless steel).
Materials:
- XRF spectrometer (ED-XRF or WD-XRF)
- A set of certified reference materials (CRMs) with known compositions covering the expected concentration ranges of all elements of interest (e.g., Fe, Cr, Ni, Mo, Mn for stainless steel) [100].
- Sample cups or holders suitable for solid metals.
- Cleaning materials (solvent, lint-free wipes).
Methodology:
- Power Up and Stabilize: Turn on the instrument and allow it to stabilize according to the manufacturer's instructions (typically 30-60 minutes).
- Set Up Analysis Method: Define the analytical program, selecting the appropriate X-ray tube voltage, current, filter, and counting time for the alloy type.
- Measure Standards: Measure each CRM in the calibration set. Ensure the sample surface is clean and properly positioned. The software will record the intensity for each element's spectral line.
- Create Calibration Curves: The instrument software will use the intensity data and the known concentrations of the CRMs to generate a calibration curve (intensity vs. concentration) for each element. Models for matrix effect correction (e.g., Fundamental Parameters) should be applied [93].
- Verify Calibration: Analyze a verification CRM (a standard not used in the calibration set). The measured values should agree with the certified values within specified tolerance limits.
- Quality Control: Routinely analyze a control sample (e.g., a verified CRM or a known in-house standard) to monitor the long-term stability and performance of the calibration [100].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential Materials for XRF Alloy Analysis
| Item | Function/Description |
|---|---|
| Certified Reference Materials (CRMs) | High-purity standards with certified elemental concentrations. Essential for accurate calibration and validation of results. Must be matrix-matched to the analyzed alloys (e.g., stainless steel CRMs for stainless steel analysis) [100] [93]. |
| Sample Preparation Tools | Cutting saws, lathes, milling machines, and abrasive papers (SiC papers of varying grits). Used to create a fresh, homogeneous, and flat surface, which is critical for reproducible and accurate analysis [100] [93]. |
| Cleaning Solvents | High-purity acetone, ethanol, or isopropanol. Used to remove grease, oils, and particulate matter from the sample surface and instrument window to prevent contamination [100]. |
| Sample Cups and Holders | Containers for holding powdered samples or small, irregularly shaped solid samples. Typically use a prolene or polycarbonate film to support the sample while allowing X-rays to pass through. |
| Instrument Calibration Standards | Specific standards, often provided by the instrument manufacturer, for daily performance verification (e.g., a SS316 standard for energy calibration and quality control) [99]. |
Technical Support Center
Frequently Asked Questions (FAQs)
Q1: What are the primary technical challenges in sample preparation for imaging mass spectrometry (IMS), and how can they be mitigated? Based on a detailed survey by the Japan Association for Imaging Mass Spectrometry (JAIMS), key challenges in sample preparation for IMS include preserving molecular integrity during collection, preventing analyte degradation during storage, and ensuring homogeneous tissue sectioning. To mitigate these issues, the survey proposes standardizing protocols for rapid freezing of tissue samples, using optimal cutting temperature (OCT) compounds that minimize interference, and storing sections at consistently low temperatures (-80°C) to maintain stability [101].
Q2: My WISER software cannot read my image data file. What should I check? WISER currently supports specific image formats. You should first verify that your file is in the .img/.hdr or TIFF/GEOTIFF/.tfw format. If your data is in a different format, you will need to convert it, as support for other formats is still under development. Furthermore, ensure that all associated files (like header files for .img data) are present and uncorrupted [37].
Q3: How can I improve the analysis of noisy Raman spectroscopy data in my pharmaceutical research? Integrating artificial intelligence, particularly deep learning, can significantly enhance the analysis of noisy Raman data. Convolutional Neural Networks (CNNs) and Long Short-Term Memory networks (LSTMs) can automatically identify complex patterns and perform feature extraction from noisy spectral data with minimal manual intervention, improving accuracy in tasks like impurity detection and component identification [102].
Q4: The performance of my imaging spectroscopy software is slow with large datasets. Are there any solutions? This is a common challenge. The developers of WISER have identified this issue and are actively working on improving efficiency with large datasets as a planned software enhancement. For commercial software like Amira, leveraging its built-in support for deep learning models and its ready-to-use Python environment can help automate and accelerate the processing and segmentation of large, complex datasets [37] [103].
Q5: What should I do if the device or instrument I am trying to add to my system is rejected? This troubleshooting step is common when integrating hardware. First, consult the official list of supported devices provided by your software or system manufacturer. If the device is listed, ensure it is in the correct pairing mode. A general procedure is to press the reset button on the device three times and then hold it for 20 seconds until an LED indicator blinks red, signaling a reboot into the correct mode [104].
Troubleshooting Guides
Table 1: Common Software Issues and Solutions
| Symptom | Possible Cause | Solution | Applicable Software/Context |
|---|---|---|---|
| Software cannot read image file | Unsupported file format | Convert data to supported formats (e.g., .img/.hdr, TIFF/GEOTIFF) [37] | WISER |
| Slow performance with large datasets | Software not optimized for large data volumes | Utilize built-in deep learning tools or await upcoming efficiency updates [37] [103] | WISER, Amira, General Imaging Spectroscopy |
| Inaccurate spectral analysis | High background noise and complex data | Apply AI-based analysis (e.g., CNN, LSTM) for automated pattern recognition [102] | Raman Spectroscopy Analysis |
| Difficulty integrating hardware/device | Device not supported or in incorrect mode | Check supported device list; reset device to correct pairing mode [104] | General Instrument Control |
Table 2: Technical Challenges in Imaging Mass Spectrometry (IMS) and Proposed Standard Methods
| Challenge Area | Specific Technical Problem | Proposed Standard Method / Realistic Approach |
|---|---|---|
| Sample Collection & Storage | Loss of molecular integrity | Standardize rapid freezing protocols and optimal storage temperatures [101] |
| Tissue Section Preparation | Inconsistent section thickness or quality | Establish standardized procedures for cryostat sectioning [101] |
| Data Analysis & Quantification | Lack of reliable quantitative analysis | Develop and validate standardized methods for data correction and quantitative calibration [101] |
| Data Reproducibility | Inter-laboratory variability | Implement and promote standardized experimental workflows across labs [101] |
Experimental Protocols and Workflows
Protocol 1: Standardized Workflow for IMS Sample Preparation
This protocol synthesizes the approaches discussed by JAIMS to enhance reproducibility in pharmaceutical research [101].
- Sample Collection: Excise tissue sample and immediately snap-freeze using liquid nitrogen or pre-chilled isopentane to preserve the spatial distribution of metabolites and drugs.
- Storage: Store the frozen tissue block at -80°C to prevent analyte degradation until sectioning.
- Tissue Sectioning:
- Use a cryostat maintained at an appropriate temperature (e.g., -20°C).
- Cut tissue sections to a standardized thickness (e.g., 5-20 µm).
- Transfer sections onto pre-cleaned, conductive glass slides or indium-tin-oxide (ITO) slides.
- Sample Preparation:
- Allow sections to thaw and dry in a desiccator to prevent moisture accumulation.
- Depending on the analysis, apply a matrix uniformly using an automated sprayer for Matrix-Assisted Laser Desorption/Ionization (MALDI)-IMS.
Protocol 2: AI-Enhanced Analysis of Raman Spectral Data
This methodology outlines the integration of AI for processing Raman spectroscopy data in drug development, as highlighted in recent reviews [102].
- Data Acquisition: Collect raw Raman spectral data from the sample(s) of interest.
- Pre-processing: Perform baseline correction and noise reduction on the raw spectra.
- Data Preparation: Split the pre-processed spectral data into training and testing sets.
- Model Training: Train a selected deep learning algorithm (e.g., a Convolutional Neural Network (CNN) for spatial feature extraction) on the training set. The model learns to correlate spectral features with specific molecular structures or conditions.
- Validation & Interpretation: Validate model predictions using the test set. Employ interpretable AI methods (e.g., attention mechanisms) to understand which spectral regions most influenced the prediction, addressing the "black box" challenge [102].
Essential Research Reagent Solutions
Table 3: Key Materials for Imaging Spectroscopy Experiments
| Item | Function/Benefit |
|---|---|
| Cryostat | Enables the production of thin, consistent tissue sections from frozen samples, which is critical for high-quality IMS and other imaging spectroscopy data [101]. |
| Optimal Cutting Temperature (OCT) Compound | An embedding medium used to support tissue during cryostat sectioning. It is vital to select OCT compounds that do not interfere with the spectral analysis [101]. |
| Conductive Glass Slides (e.g., ITO slides) | Essential for IMS techniques like MALDI, as they provide a conductive surface required for the ionization process [101]. |
| Matrix Compounds (e.g., CHCA, DHB) | Used in MALDI-IMS to co-crystallize with the sample, absorb laser energy, and promote the desorption and ionization of analytes for mass spectrometry analysis [101]. |
| Deep Learning Models (CNNs, LSTMs) | AI tools that function as "software reagents" to automatically and efficiently process, denoise, and interpret complex spectral data, overcoming limitations of manual analysis [102]. |
Workflow and Relationship Diagrams
Software Selection Workflow
AI Raman Analysis Workflow
For researchers, scientists, and drug development professionals, selecting the right spectroscopy software is a critical decision that directly impacts data integrity, analytical efficiency, and compliance. This guide provides a structured framework for benchmarking software capabilities, supported by troubleshooting guides and FAQs to address common experimental challenges.
Key Evaluation Criteria for Spectroscopy Software
When selecting spectroscopy software, a systematic evaluation based on the following criteria is essential to ensure it meets both current and future needs.
1.1 Core Functional Capabilities Modern spectroscopy software should offer a comprehensive suite of functionalities that span the entire data lifecycle, from acquisition to reporting. Key aspects to evaluate include:
- Data Processing and Analysis: Look for robust tools for peak picking, integration, baseline correction, and spectral deconvolution. Advanced software incorporates AI-powered algorithms for tasks like automatic peak picking and baseline correction, enhancing accuracy and reproducibility [105].
- Multi-Technique and Vendor-Neutral Support: The ability to handle data from multiple spectroscopic techniques (e.g., NMR, LC/MS, IR, Raman) and different instrument vendors (e.g., Bruker, Agilent, Thermo Fisher) within a single platform streamlines workflows and reduces the learning curve [106].
- Reporting and Publication Tools: Software should facilitate the creation of comprehensive, publication-ready reports. The ability to customize report templates and automatically assemble annotated spectra, chromatograms, and peak tables is a significant time-saver [106].
1.2 User Experience and Technical Performance
- User Interface (UI) and Usability: The interface should be intuitive, with a shallow learning curve for beginners yet powerful enough for experts. Features like drag-and-drop data loading, automated processing workflows, and clear visualization modes are key indicators of good UI design [106] [105].
- Data Management and Integration: Assess how the software handles data storage, retrieval, and security. Furthermore, its ability to integrate with existing laboratory infrastructure, such as Laboratory Information Management Systems (LIMS) and electronic lab notebooks (ELNs), is crucial for data integrity and workflow efficiency [107].
- Deployment and Accessibility: Software can be deployed on-premise or via the cloud. On-premise solutions are often preferred for direct control over sensitive data and compliance, while cloud-based platforms facilitate remote access, collaboration, and scalability [1].
1.3 Support and Compliance
- Technical Support and Training: The availability of responsive, expert technical support and comprehensive training resources is vital for resolving issues quickly and minimizing downtime.
- Regulatory Compliance: For pharmaceutical and other regulated industries, software must support compliance with standards like 21 CFR Part 11. This includes features for audit trails, electronic signatures, and data integrity protection [108] [109].
Table 1: Software Evaluation Criteria at a Glance
| Category | Key Criteria | Questions to Ask |
|---|---|---|
| Core Functionality | Data Processing, Multi-technique Support, Reporting | Does it support all our instrument data formats? Can it create publication-ready reports? |
| User Experience | Interface Usability, Workflow Automation, Data Management | Is the interface intuitive for all user levels? Can we automate routine analysis? |
| Technical Performance | Deployment Model (On-premise/Cloud), Speed, Scalability | Does it meet our data security and remote access needs? How does it perform with large datasets? |
| Support & Compliance | Technical Support, Regulatory Features (e.g., 21 CFR Part 11), Validation | What support and training are available? Does it have built-in audit trails and e-signatures? |
Essential Research Reagent Solutions
The following reagents and materials are fundamental for a wide range of spectroscopy-based experiments in drug development and research.
Table 2: Key Research Reagents and Materials
| Reagent/Material | Primary Function in Spectroscopy |
|---|---|
| Deuterated Solvents (e.g., D₂O, CDCl₃) | Provides a non-interfering signal background for NMR spectroscopy [106]. |
| Proteinase K | Digests proteins and removes contamination in nucleic acid samples for accurate UV-Vis analysis [108]. |
| iTRAQ / TMT Reagents | Enables multiplexed protein quantification in mass spectrometry-based proteomics [110]. |
| PNIPAM Polymer | Used in material science research, often studied via UV-Vis to monitor temperature-dependent aggregation of nanoparticles [108]. |
| NIST Standard Reference Materials | Provides certified standards for instrument calibration and method validation across various spectroscopic techniques [110]. |
Troubleshooting Guides and FAQs
This section addresses specific, common issues users might encounter during their experiments.
Data Processing and Analysis
Q1: My NMR spectrum has a poor signal-to-noise ratio. What are the common causes and solutions?
- Cause 1: Insufficient scan numbers. Solution: Increase the number of scans (transients) to improve signal averaging.
- Cause 2: Incorrect probe tuning or shimming. Solution: Ensure the NMR probe is properly tuned and matched for your solvent, and that the magnetic field is correctly shimmed for optimal resolution.
- Cause 3: Sample-related issues (e.g., low concentration, paramagnetic impurities). Solution: Confirm sample concentration and purity. Filter or re-prepare the sample if necessary.
Q2: The software's automated peak picking for my Raman spectrum is inaccurate, missing small peaks or selecting noise. How can I improve this?
- Solution 1: Manually adjust the sensitivity threshold. Most software allows you to set a minimum intensity or signal-to-noise ratio for peak detection.
- Solution 2: Use advanced algorithms. If available, leverage AI-powered or Global Spectral Deconvolution (GSD) tools, which are designed to separate overlapping signals and improve picking accuracy [105].
- Solution 3: Apply smoothing or baseline correction first. Pre-processing the spectrum to reduce noise and correct the baseline can significantly improve automated peak picking results.
Q3: How do I handle and analyze data from multiple analytical techniques (e.g., NMR, MS, IR) for the same sample?
- Solution: Utilize a vendor-neutral, multi-technique software platform. Tools like Spectrus Processor allow you to assemble all related analytical data in a single dashboard, enabling you to review complementary data together for faster and more confident structural elucidation or verification [106].
Software Operation and Workflow
Q4: I need to ensure my UV-Vis software is compliant with 21 CFR Part 11. What key features should I verify?
- Solution: The software must have features for access control (unique user logins), audit trails (a secure, time-stamped record of all user actions), and electronic signatures [108]. Consult the software vendor's documentation for a specific compliance module or configuration guide.
Q5: My data processing workflow is repetitive and time-consuming. Can I automate it?
- Solution: Yes. Many advanced software packages, like Mnova, support the use of scripting tools (e.g., Python) and processing templates. You can record a series of steps and save them as a template to be applied automatically to new datasets, ensuring consistency and saving valuable time [105].
Q6: What is the benefit of using Multivariate Data Analysis (MVDA) in my spectroscopy work?
- Solution: MVDA, using methods like Principal Component Analysis (PCA), allows you to analyze multiple variables from complex datasets simultaneously. In spectroscopy, this is powerful for identifying patterns, classifying samples, detecting outliers, and understanding how different factors influence your spectral data, which is particularly useful in Process Analytical Technology (PAT) and omics studies [109].
Experimental Protocols and Workflows
Protocol: Conducting a Principal Component Analysis (PCA) on Spectral Data
This protocol outlines the steps to perform PCA, a common MVDA technique, using a series of spectra (e.g., NIR, Raman) to classify samples or identify spectral patterns.
1. Objective: To reduce the dimensionality of a spectral dataset and visualize natural groupings or trends among samples. 2. Materials and Software: - Spectral dataset (multiple spectra from different samples). - MVDA software (e.g., SIMCA, or built-in chemometrics tools in platforms like Mnova). 3. Methodology: - Step 1: Data Assembly and Export. Collect all spectra and ensure they are in a compatible format for your MVDA software. Export the data, typically as a matrix where rows represent samples and columns represent variables (e.g., intensity at each wavelength/wavenumber). - Step 2: Data Pre-processing. Load the data into the MVDA software. Apply necessary pre-processing steps to remove unwanted variance. Common methods include: - Baseline Correction: To remove baseline shifts. - Normalization: To correct for differences in overall signal intensity between samples. - Mean Centering: A standard step in PCA that makes the model focus on variance rather than absolute values. - Step 3: Model Generation. Select the PCA algorithm and generate the model. The software will calculate the principal components (PCs), which are new variables that capture the maximum variance in the data. - Step 4: Model Interpretation. Analyze the output: - Scores Plot: This scatter plot (e.g., PC1 vs. PC2) shows how your samples relate to each other. Clustered samples have similar spectral properties. - Loadings Plot: This plot explains what original variables (wavelengths/wavenumbers) are responsible for the patterns seen in the scores plot. Peaks in the loadings indicate spectral regions that contribute most to the separation of samples. 4. Troubleshooting: - No Clear Grouping: This may indicate no inherent chemical differences between sample groups, or that pre-processing needs optimization. - Model is Complex (Too many PCs needed): Explore other pre-processing methods or consider using supervised multivariate methods like PLS.
Workflow Diagram: Troubleshooting Poor Spectral Quality
The following diagram outlines a logical, step-by-step workflow for diagnosing and resolving common issues that lead to poor-quality spectra, applicable to various spectroscopic techniques.
Diagram 1: A logical workflow for troubleshooting poor spectral quality, guiding users from problem identification to potential solutions.
Benchmarking spectroscopy software requires a balanced consideration of technical capabilities, user-centric design, and robust support structures. By applying the structured criteria outlined here—from core functionality and UI to integration and compliance—research teams can make informed decisions that enhance their data analysis capabilities. Furthermore, leveraging troubleshooting guides and established experimental protocols helps overcome common practical challenges, ensuring data quality and accelerating the drug development pipeline. The integration of AI, cloud computing, and advanced MVDA tools will continue to shape the future of spectroscopy software, making the adoption of a rigorous evaluation framework more important than ever.
Conclusion
The integration of advanced software tools is fundamentally transforming spectroscopy from a data collection technique into a powerful, intelligent platform for discovery and validation in biomedical research. Key takeaways highlight the non-negotiable necessity of rigorous method validation, the critical productivity gains from optimized sample preparation workflows, and the growing impact of AI and portable technologies. Looking forward, the continued convergence of spectroscopy with other analytical techniques, the expansion of open-source software platforms, and the deepening application of machine learning will further accelerate drug development, enable more sophisticated diagnostic methods, and push the boundaries of personalized medicine. For researchers and drug development professionals, staying abreast of these trends is essential for maintaining a competitive edge and achieving regulatory success.
References
- 1. Spectroscopy Software Market: 2025-2034 Growth ... [https://www.gminsights.com/industry-analysis/spectroscopy-software-market]
- 2. Spectroscopy Software Market Report 2025 [https://www.researchandmarkets.com/reports/6168438/spectroscopy-software-market-report?srsltid=AfmBOoqsOmiGjaYg9g_AeuRchEPk6LKrbfP8Hp3-wp6OBtV_gfaeOvsx]
- 3. Spectroscopy Software Market Opportunity, Growth Drivers ... [https://marketpublishers.com/report/life_sciences/healthcare/spectroscopy-software-market-opportunity-growth-drivers-industry-trend-analysis-n-forecast-2025-v-2034.html]
- 4. Spectroscopy Software Market Size & Share 2025-2032 [https://www.360iresearch.com/library/intelligence/spectroscopy-software]
- 5. spectroscopy software market By Type, By Application, ... [https://www.marketsizeandtrends.com/report/spectroscopy-software-market/]
- 6. Spectroscopy Software Market Opportunity Landscape | AI ... [https://www.linkedin.com/pulse/spectroscopy-software-market-opportunity-landscape-hymmc/]
- 7. 2025 Review of Spectroscopic Instrumentation [https://www.spectroscopyonline.com/view/2025-review-of-spectroscopic-instrumentation]
- 8. 2025 guide to pharmaceutical software [https://www.qualio.com/blog/pharmaceutical-software]
- 9. 15 Essential Lab Software Every Scientist Should Know ... [https://www.scispot.com/blog/essential-lab-software-for-every-lab]
- 10. FT-IR Troubleshooting Guide: How to Solve Common ... [https://www.spectroscopyonline.com/view/ftir-troubleshooting-guide-how-to-solve-common-problems-in-infrared-spectroscopy]
- 11. Xcalibur Data Acquisition and Interpretation Software [https://www.thermofisher.com/us/en/home/industrial/mass-spectrometry/liquid-chromatography-mass-spectrometry-lc-ms/lc-ms-software/lc-ms-data-acquisition-software/xcalibur-data-acquisition-interpretation-software.html]
- 12. Technical Support | Lambda Scientific [https://lambdasci.com/technical-support]
- 13. Real-Time PCR and Digital PCR Instruments Support Center [https://www.thermofisher.com/us/en/home/technical-resources/technical-reference-library/real-time-digital-PCR-instruments-support-center.html]
- 14. Spectroscopy Software [https://www.horiba.com/usa/scientific/products/detail/action/show/Product/spectroscopy-software-1628/]
- 15. Color Measurement Software, Chromaticity, Cary WinUV [https://www.agilent.com/en/product/molecular-spectroscopy/uv-vis-uv-vis-nir-spectroscopy/uv-vis-uv-vis-nir-software/cary-winuv-color-measurement-software?srsltid=AfmBOopuEzzbQqzZ1uOirtlQGLKsmwF3Z2shWA8PGo-ISwejBXuSOjM7]
- 16. Recent Research in Chemometrics and AI for ... [https://www.spectroscopyonline.com/view/recent-research-in-chemometrics-and-ai-for-spectroscopy-part-i-foundations-definitions-and-the-integration-of-artificial-intelligence-in-chemometric-analysis]
- 17. Smarter Spectroscopy With a New Machine Learning ... [https://www.spectroscopyonline.com/view/smarter-spectroscopy-with-a-new-machine-learning-approach-to-estimate-prediction-uncertainty]
- 18. Suggestions for improving the visualization of magnetic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7481722/]
- 19. Exploring the Spectrum of Analytical Techniques for ... [https://www.spectroscopyonline.com/view/exploring-the-spectrum-of-analytical-techniques-for-material-characterization]
- 20. Resolving Analytical Challenges in Pharmaceutical ... [https://www.spectroscopyonline.com/view/resolving-analytical-challenges-in-pharmaceutical-process-monitoring-using-multivariate-analysis-methods-applications-in-process-understanding-control-and-improvement]
- 21. Machine learning spectroscopy to advance computation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12590498/]
- 22. Spectroscopy: Interpreting Measurement Data [https://www.yokogawa.com/us/library/resources/media-publications/spectroscopy-interpreting-measurement-data/]
- 23. Cloud vs On-premise Security: 6 Critical Differences [https://www.sentinelone.com/cybersecurity-101/cloud-security/cloud-vs-on-premise-security/]
- 24. On-Premise vs Cloud Software: Which is better in 2025? [https://usersnap.com/blog/comparison-of-cloud-vs-on-premise-enterprise-software/]
- 25. 10 Benefits of Moving Your Software to the Cloud in 2025 [https://www.graphon.com/blog/10-benefits-of-moving-your-software-to-the-cloud]
- 26. What are the Pros and Cons of Cloud Computing: 2025 ... [https://clearfuze.com/blog/pros-and-cons-of-cloud-computing/]
- 27. Analytical Instrument Vendors Post Strong Q2 2025 Growth ... [https://www.chromatographyonline.com/view/analytical-instrument-vendors-q2-2025-pharma-chemical-growth]
- 28. How Cloud Computing Flexibility Fuels Business Innovation [https://totalit.com/how-cloud-computing-flexibility-fuels-business-innovation/]
- 29. Preventing Breaches: Best Practices in Data Security - Spectra [https://www.spectraintegration.com/preventing-breaches-best-practices-in-data-security/]
- 30. Cloud vs On-Premise Infrastructure: Comparison Guide [https://hypersense-software.com/blog/2025/07/31/cloud-vs-on-premise-infrastructure-guide/]
- 31. FTIR | FTIR Analysis Software | Thermo Fisher Scientific - US [https://www.thermofisher.com/us/en/home/industrial/spectroscopy-elemental-isotope-analysis/molecular-spectroscopy/fourier-transform-infrared-spectroscopy/software.html]
- 32. Diagnosing & Fixing Performance Issues in Mini Portable ... [https://secuzoan.com/news/1168.html]
- 33. Welcome to Ramansystems - Troubleshooting [http://ramansystems.com/english/troubleshooting.htm]
- 34. Design and Experiment of a Portable Near-Infrared ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10610571/]
- 35. Portable Handheld Spectrometers | Malvern Panalytical [https://www.malvernpanalytical.com/en/products/category/handheld-spectrometers]
- 36. Vernier Spectral Analysis® [https://www.vernier.com/product/spectral-analysis/]
- 37. WISER software for analysis of imaging spectroscopy data ... [https://ehlmann.caltech.edu/wiser/index.html]
- 38. Pharmaceutical Quality Control Labs (7/93) [https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/inspection-guides/pharmaceutical-quality-control-labs-793]
- 39. CHROMtalks 2025: Mastering Hyphenated Method ... [https://www.chromatographyonline.com/view/chromtalks-2025-mastering-hyphenated-method-development]
- 40. Tools shaping drug discovery and development - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10903431/]
- 41. How Making Informed Decisions With Data Visualization ... [https://www.labmanager.com/how-making-informed-decisions-with-data-visualization-works-1785]
- 42. A Comparative Analysis of Cheminformatics Platforms [https://intuitionlabs.ai/articles/cheminformatics-platforms-comparison]
- 43. Hybrid Analytical Techniques: GC-MS, LC-MS, GC-IR, LC- ... [https://www.alwsci.com/news/hybrid-analytical-techniques-gc-ms-lc-ms-gc-69494187.html]
- 44. A Guide To Troubleshooting Mass Spectrometry [https://gentechscientific.com/a-guide-to-troubleshooting-mass-spectrometry/]
- 45. HPLC Troubleshooting Guide [https://scioninstruments.com/us/blog/hplc-troubleshooting-guide-2/]
- 46. Advancements and Emerging Techniques in Mass ... [https://www.chromatographyonline.com/view/advancements-and-emerging-techniques-in-mass-spectrometry-a-comprehensive-review]
- 47. Chromatography and Mass Spectrometry: Evolving ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12345980/]
- 48. Guide to QCL Microscopy [https://www.bruker.com/en/products-and-solutions/infrared-and-raman/ft-ir-microscopes/what-is-QCL-microscopy.html]
- 49. HYPERION II FT-IR and QCL Microscope [https://www.bruker.com/en/products-and-solutions/infrared-and-raman/ft-ir-microscopes/hyperion-ii-ft-ir-and-qcl-microscope.html]
- 50. Common Problems with FT-IR Instruments and How to ... [https://www.spectroscopyonline.com/view/common-problems-with-ft-ir-instruments-and-how-to-avoid-them]
- 51. Novel spectrometry Market Report 2025 [https://www.researchandmarkets.com/reports/6035525/novel-spectrometry-market-report?srsltid=AfmBOopSxW6CVx9Wz4PCXowaYAY43a-uXWlN8SyU94DSDDMQfZdGf4gv]
- 52. Novel Spectrometry Platforms Market Report 2025 (Global Edition) [https://www.cognitivemarketresearch.com/novel-spectrometry-platforms-market-report]
- 53. Novel Spectrometry Platforms Market Trends Forecast, 2032 [https://www.coherentmi.com/industry-reports/novel-spectrometry-platforms-market]
- 54. 'Total Workflow' Sample Preparation Approach to optimize ... [https://www.milestonesrl.com/homepage-en/total-workflow-sample-prep-approach-to-optimize-elemental-analysis]
- 55. Tips and tricks for successful Mass spec experiments [https://www.ptglab.com/news/blog/tips-and-tricks-for-successful-mass-spec-experiments/?srsltid=AfmBOopwcmtdsa2-id1do4a5RANTpitIqEi426Iq4f8r6YHjcSr-BxLl]
- 56. What Causes Errors In Ir Spectroscopy? Master Sample ... [https://kindle-tech.com/faqs/what-causes-errors-in-ir-spectroscopy?srsltid=AfmBOopZH0mOmHnyjnRW6WMUVIi9X9FAB87yb9l_-3zkec5CbM9GgBRG]
- 57. - A TEEM Spectroscopy [https://www.horiba.com/rus/scientific/products/fluorescence-spectrometers/a-teem/]
- 58. Using A-TEEM Molecular Fingerprinting for Pharmaceutical ... [https://www.azom.com/article.aspx?ArticleID=22215]
- 59. Compliance Solutions for Using A-TEEM Spectroscopy in ... [https://www.spectroscopyonline.com/view/compliance-solutions-for-using-a-teem-spectroscopy-in-gmp-labs]
- 60. Long-term device stability for Raman spectroscopy [https://pubs.rsc.org/en/content/articlehtml/2025/an/d5an00255a]
- 61. IR vs | Separation Science Raman Spectroscopy [https://www.sepscience.com/ir-vs-raman-spectroscopy-9516]
- 62. Biopharma and Pharma [https://www.horiba.com/int/life-science/applications/biopharma-and-pharma/]
- 63. Optical Emission Spectrometer System Common Vacuum ... Failures [https://www.jinyibo.com/blog/optical-emission-spectrometer-vacuum-system-common-failures-and-solutions-vacuum-pump-cannot-pump-down_b63]
- 64. Liquid Ring Vacuum : Expert Pump Troubleshooting Guide [https://www.vacculex.com/blogs/liquid-ring-vacuum-pump-troubleshooting/]
- 65. Mass Spectrometry 101 Troubleshooting [https://www.numberanalytics.com/blog/mass-spectrometry-troubleshooting-guide]
- 66. The Importance of Routine Maintenance in ICP-MS [https://www.spectroscopyonline.com/view/the-importance-of-routine-maintenance-in-icp-ms]
- 67. Five Simple Rules for Fiber and Probe Maintenance [https://www.oceanoptics.com/blog/fiber-and-probe-maintenance/]
- 68. Quality control and removal of technical variation of NMR ... [https://www.nature.com/articles/s41597-023-01949-y]
- 69. Pre-processing spectroscopic data: for good or ill? [https://www.spectroscopyeurope.com/td-column/pre-processing-spectroscopic-data-good-or-ill]
- 70. Data Quality and Data Integrity Are the Same, Right? Wrong! [https://www.spectroscopyonline.com/view/data-quality-and-data-integrity-are-same-right-wrong]
- 71. Beyond the Paradigm: Combining Mass Spectrometry and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5448308/]
- 72. Best Practices for Raman Spectroscopy Data Analysis [https://www.sartorius.com/en/knowledge/science-snippets/best-practices-for-raman-spectroscopy-data-analysis-1017174]
- 73. Errors in Spectrophotometry and Calibration Procedures to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5293527/]
- 74. Simple Spectrometer System, Simple Validation? [https://www.spectroscopyonline.com/view/simple-spectrometer-system-simple-validation-]
- 75. Troubleshooting Common Spectrometer Issues [https://verichek.net/troubleshooting-common-spectrometer-issues.html]
- 76. Maintenance & calibration of optical instruments [https://www.opticalscientific.com/maintenance-calibration-of-optical-instruments/]
- 77. Planned Maintenance | Thermo Fisher Scientific - US [https://www.thermofisher.com/us/en/home/products-and-services/services/instrument-qualification-services/instrument-services/planned-maintenance.html]
- 78. OptiCentric® - Lens centration and alignment [https://www.trioptics.us/products/opticentric-lens-centration-and-alignment]
- 79. How Automation Overcomes Variability & Limitations in ... [https://dispendix.com/blog/how-automation-overcomes-variability-limitations-in-hts-troubleshooting]
- 80. The Benefits of Automation in High Throughput Screening [https://dispendix.com/blog/benefits-of-automation-in-high-throughput-screening]
- 81. A Fully Automated High-Throughput Flow Cytometry ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6055113/]
- 82. Best Practices Analytical Method Validation [https://www.numberanalytics.com/blog/analytical-method-validation-best-practices]
- 83. Best Practices For Successful Method Validation [https://lgmpharma.com/blog/best-practices-for-successful-method-validation/]
- 84. Understanding the Four Types of Analytical Method Validation [https://www.oxford-analytical.co.uk/understanding-the-four-types-of-analytical-method-validation/]
- 85. Parameters: An Overview Analytical Method Validation [https://www.linkedin.com/pulse/analytical-method-validation-parameters-overview-fokhrul-abedin-glhrf]
- 86. Common Mistakes in Method Validation and How to Avoid ... [https://www.linkedin.com/pulse/common-mistakes-method-validation-how-avoid-them-part-oona-mcpolin-1]
- 87. Spectrophotometer Troubleshooting [https://www.hinotek.com/spectrophotometer-troubleshooting/]
- 88. Detection limit [https://en.wikipedia.org/wiki/Detection_limit]
- 89. Limit of Blank, Limit of Detection and Limit of Quantitation [https://pmc.ncbi.nlm.nih.gov/articles/PMC2556583/]
- 90. The Limit of Detection [https://www.chromatographyonline.com/view/limit-detection]
- 91. Validation of analytical methods and detection limits in ... [https://www.sciencedirect.com/science/article/pii/S0969806X24009745]
- 92. Managing the Analytical Life-Cycle for Biotechnology ... [https://www.bioprocessintl.com/bioanalytical-methods/managing-the-analytical-life-cycle-for-biotechnology-products]
- 93. X-Ray Fluorescence (XRF): Theory, Practice and ... [https://www.technologynetworks.com/analysis/articles/x-ray-fluorescence-xrf-theory-practice-and-applications-396212]
- 94. What is X-ray Fluorescence (XRF) [https://www.horiba.com/usa/scientific/technologies/energy-dispersive-x-ray-fluorescence-ed-xrf/what-is-x-ray-fluorescence-xrf/]
- 95. EDXRF Vs WDXRF: Choosing the Right Chemical Analysis ... [https://www.thermofisher.com/blog/materials/edxrf-vs-wdxrf-choosing-the-right-chemical-analysis-method/]
- 96. EDXRF (energy dispersive X-ray fluorescence ... [https://rigaku.com/products/xrf-spectrometers/edxrf/technique]
- 97. Energy Dispersive XRF (ED-XRF) VS. Wavelength ... [https://www.vrxrf.com/energy-dispersive-xrf-ed-xrf-vs-wavelength-dispersive-xrf-wd-xrf-which-one-is-right-for-your-industry/]
- 98. Comparing WDXRF and EDXRF - Which Spectrometer is ... [https://www.drawellanalytical.com/comparing-wdxrf-and-edxrf-which-spectrometer-is-right-for-your-needs/]
- 99. XRF Troubleshooting [https://www.portablescientific.com.au/blog/xrf-troubleshooting/]
- 100. Avoiding Common Pitfalls in XRF Analysis [https://www.vrxrf.com/avoiding-errors-in-xrf-analysis/]
- 101. Current Status of Imaging Mass Spectrometry in Drug ... [https://www.sciencedirect.com/science/article/abs/pii/S1347436725004355]
- 102. AI-Powered Raman Spectroscopy Signals New Era for ... [https://www.spectroscopyonline.com/view/ai-powered-raman-spectroscopy-signals-new-era-for-drug-development-and-disease-diagnosis]
- 103. Amira Software for drug discovery [https://www.thermofisher.com/us/en/home/electron-microscopy/products/software-em-3d-vis/drug-discovery-design-manufacturing.html]
- 104. Troubleshooting - Wiser Home [https://www.productinfo.schneider-electric.com/wiser_home/wiser-home-sug-iberic/English/System%20User%20Guide_Wiser_Home%20(Iberic)_DD00528175.xml/$/SUG_Home-Troubleshooting-FB340499]
- 105. Mnova NMR Software for 1D and 2D NMR Data [https://mestrelab.com/main-product/nmr]
- 106. Software for NMR, Chromatography, MS & more | Spectrus ... [https://www.acdlabs.com/products/spectrus-platform/spectrus-processor/]
- 107. Spectroscopy Software Market Report 2025 [https://www.researchandmarkets.com/reports/6168438/spectroscopy-software-market-report?srsltid=AfmBOoqemsKns_H2Ra7Yoc08ePbJmcqzhiU77DP9UE8Upa64W-RF24LX]
- 108. UV-Vis & UV-Vis-NIR Software - Molecular Spectroscopy [https://www.agilent.com/en/product/molecular-spectroscopy/uv-vis-uv-vis-nir-spectroscopy/uv-vis-uv-vis-nir-software?srsltid=AfmBOopkScTLEyQ_hRAAXzkY_uAP2EotY8RR9LL0stE8DoKquG51dTWg]
- 109. Multivariate Data Analysis Software [https://www.sartorius.com/en/products/process-analytical-technology/data-analytics-software/mvda-software]
- 110. List of mass spectrometry software [https://en.wikipedia.org/wiki/List_of_mass_spectrometry_software]